Review Article
Use of MicroRNAs to Screen for Colon Cancer

Farid E Ahmed1*, Nancy C Ahmed2, Mostafa Gouda2 and Chris Bonnerup3
1GEM Tox Labs, Institute for Research in Biotechnology, 2905 South Memorial Drive, Greenville, NC 27834, USA
2National Research and Development Center for Egg Processing, College of Food Science and Technology, Huazhong Agricultural University, Wuhan, Hubei, PR, China
3Department of Physics, East Carolina University, Greenville, NC, 27834, USA
*Address for Correspondence: Farid E Ahmed, GEM Tox Labs, Institute for Research in Biotechnology, 2905 South Memorial Drive, Greenville, NC 27834, USA, Tel: +1 (252) 864-1295; Email: [email protected]
Dates: Submitted: 15 July 2017; Approved: 30 August 2017; Published: 31 August 2017
How to cite this article: Ahmed FE, Ahmed NC, Gouda M, Bonnerup C. Use of MicroRNAs to Screen for Colon Cancer. Insights Biol Med. 2017; 1: 045-074. DOI: 10.29328/journal.ibm.1001006
Copyright License: © 2017 Ahmed FE, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Bioinformatics; Diagnosis; Histopathology; Microarrays; QC, RNA, RT-qPCR, Statistics
Abbreviations: ACS: American Cancer Society; ANOVA: Analysis of Variance by Statistics; APC: Adenomatous Polyposis Coli Gene; CA: Carcinoembryonic Antigen; CC: Colon Cancer; CP: Comparative Cross Point; CRC: Colorectal Cancer; dMMR: Defective DNA Mismatch Repair; DNMTs: DNA Methylation Enzymes; CRC: Colorectal Cancer; DAVID: Bioinformatics Tool Referring to Database for Annotation, Visualization and Integrated Discovery; E: Efficiency of the polymerase chain reaction; EDTA: Ethylenediminetetraacetic Acid; E-method, another name for the comparative cross point method for polymerase chain reaction quantification; FOBT: Fecal Occult Blood Test; GESS: Gene Expression Statistical System; GMOs: Genetically Modified Organisms; IBD: Inflammatory Bowel Disease; IHC: Immunohistological; LC: Light Cycler Instrument; LCM: Laser Capture Microdissection; MIQUE, guidelines on reporting qPCR data known as minimum information for publication of quantitative real-time PCR expression; NCI-EORTC: National Cancer Institute and the European Organization for Research and Treatment of Cancer; NCSS: Statistical Software; NF1A: Nuclear factor 1A-type protein; pMMR: Proficient In Dna Mismatch Repair; PRoBE: Epidemiological Experimental Random Design; QC: Quality Control; qPCR: Quantitative polymerase chain reaction; 18s rRNA: Ribosomal Ribonucleic Acid; RT: Reverse Transcription reaction; SYBR Green, an asymmetrical canine dye for nucleic acids staining; TNM staging, a cancer staging notation system; TPC: Test Performance Characteristics; UC: Ulcerative Colitis; UTR, the 3’ Untranslated region of target messenger RNA
Abstract
Colon cancer (CC) screening is important for diagnosing early stage for malignancy and therefore potentially reduces mortality from this disease because the cancer could be cured at the early disease stage. Early detection is needed if accurate and cost effective diagnostic methods are available. Mortality from colon cancer is theoretically preventable through screening. The Current screening method, the immunological fecal occult blood test, FOBTi, lacks sensitivity and requires dietary restriction, which impedes compliance. Moreover colonoscopy is invasive and costly, which decreases compliance, and in certain cases could lead to mortality. Compared to the FOBT test, a noninvasive sensitive screen that does not require dietary restriction would be more convenient. Colonoscopy screening is recommended for colorectal cancer (CRC). Although it is a reliable screening method, colonoscopy is an invasive test, often accompanied by abdominal pain, has potential complications and has high cost, which have hampered its application worldwide.
A screening approach that uses the relatively stable and nondegradable microRNA molecules when extracted from either the noninvasive human stool, or the semi-invasive blood samples by available commercial kits and manipulated thereafter, would be more preferable than a transcriptomic messenger (m)RNA-, a mutation DNA-, an epigenetic-or a proteomic-based test. That approach utilizes reverse transcriptase (RT), followed by a modified quantitative real-time polymerase chain reaction (qPCR). To compensate for exosomal miRNAs that would not be measured, a parallel test could be performed on stool or plasma’s total RNAs, and corrections for exosomal loss are made to obtain accurate results. Ultimately, a chip would be developed to facilitate diagnosis, as has been carried out for the quantification of genetically modified organisms (GMOs) in foods. The gold standard to which the miRNA test is compared to is colonoscopy. If laboratory performance criteria are met, a miRNA test in human stool or blood samples based on high throughput automated technologies and quantitative expression measurements currently employed in the diagnostic clinical laboratory, would eventually be advanced to the clinical setting, making a noticeable impact on the prevention of colon cancer.
Introduction
Colon cancer is a disease that is different from rectal cancer [1]. In this article, we have focused on colon cancer (CC) screening, which is the process of looking for the disease in people showing no symptoms for malignancy [1,2]. Regular screening can detect colon cancer at its early stages, when it is most likely curable, because if growing polyps are observed, they can be removed before they have a chance to develop into a full-blown cancer [3]. It should be stressed, however, that none of the tests currently employed on the market is optimal, and they also have poor rates in certain populations.
Tests for colon cancer screening fall into two categories [4]: a) tests that detect both polyps and cancer, and looks at the structure of the colon to find any abnormalities. This is carried out with an x-ray either after ingesting a contrasting liquid, followed by inserting a scope into the rectum (flexible sigmoidoscopy, capsule endoscopy, double contrast barium enema), or in other tests that employs special x-ray imaging such, as CT colonography (virtual colonoscopy). These tests although are invasive, they allow for the removal of polyps when observed, and thus have a role in colon cancer prevention, or b) in vitro tests that generally looks at the genetic material (DNA or RNA) in a non-invasive excrement (stool) or in a semi-invasive body fluid (blood), so that tests with high sensitivity and specificity, capable to function as an acceptable screen for this preventable cancer (e.g., guaiac- and immunological-based FOBTs, and molecular DNA tests in either stool and blood) are developed. These in vitro tests are less invasive and are easier to carry out, but many of them have low sensitivity for polyps’ detection, unless they are further developed and refined [1,4,6,10-15]. Therefore, much effort and expense have been spent during the last 20 years to develop acceptable non-invasive tests [5-14]. These tests can be used when people exhibit symptoms of colon cancer, or other digestive diseases to check on the progression of the anomalies.
Methods for colon cancer screening
When recommended, screening often begins with fecal occult blood test, FOBT, which is blood that cannot be seen with the naked eye in stool [3,5,15]. Many CRCs bleed into the intestinal lumen because blood vessels at the surface of large polyps or cancer are fragile and can easily be damaged by the passage of feces, releasing a small amount of blood into the stool, and FOBT can detect the invisible occurrence of blood in stool by a chemical reaction. The test cannot tell if blood is from the colon or from other parts of the digestive tract (e.g., stomach). Although polyps and cancers cause blood in stool, other causes of bleeding are ulcers, hemorrhoids, diverticulosis (tiny pouches that form at weak spots in colon wall), or IBDs (colitis) [11]. Nonetheless, as blood passes through the intestine, it becomes degraded, and depending upon the site at which the hemorrhage occurs, blood detected in the stool by FOBT will vary. Thus, FOBT alone has a limited ability to decrease mortality, as 67-85% of colon cancer patients who undergone FOBT died from the disease, indicating that its detection does not occur early enough to maximally affect the overall outcome of the disease, and therefore FOBT is not a sensitive test since it misses many early stage cancers and adenomas. Moreover, guaiac FOBT test requires patients to change their diet before testing, avoid nonsteroidal anti-inflammatory drugs (NSAIDs) like ibuprofen (Advil), naproxen (Aleve) or aspirin (> 1 adult aspirin, 325 mg/day) for 7 days before testing as they cause bleeding, although Tylenol® can be taken as needed, vitamin C in excess of 250 mg/day from all sources, and red meats (beef, lamb or liver) for 3 days before testing, because components of blood in meat could give false positive results [1,4-6,15-17]. The procedure requires multiple tests to be repeated every year, potentially reducing compliance [18]. Moreover, if the test finds blood, a colonoscopy will be required to look for the source (American Cancer Society, http://ww.cancer.org).
A more recent test than the traditional guaiac is fecal immunochemical test (FIT) or (iFOBT), which reacts to part of the human hemoglobin protein found in red blood cells. This test is easier to use than guaiac FOBT because it requires no drug or dietary restrictions, and it is less likely to react to bleeding from parts of the upper digestive tract (e.g., stomach) [4,16]. Because like guaiac FOBT, the FIT will not react to a non-bleeding tumor [17,18], multiple stool samples are required for testing, and if results are positive, a colonoscopy will also be necessary.
In contrast to FOBTs, minimally invasive procedures could detect neoplastic lesions. Since > 60% of early lesions seem to arise in the rectosigmoid areas of the large intestine, rigid sigmoidoscopy, about 60 cm long, which can only see half the colon, has been routinely used in the past for screening [19]. Recently, however, there has been an increase in the number of lesions arising from more proximal lesions of the colon [6,20-23], requiring the use of flexible, fiber optic sigmoidoscopies. Although these methods offer a means of removing neoplastic polyps, they still leave undetected all lesions that are beyond the reach of the scope (estimated to be between 25 and 34%) [19].
Double-contrast barium enema (DCBE), also referred to as air-contrast barium enema, or a barium enema with air contrast and sometimes known as lower GI series, is basically a type of an x-ray test in which a chalky liquid (barium sulfate) and air is used to outline the inner part of the colon and rectum to look for abnormal areas on x-rays [1-5]. A clear liquid diet is taken for a day or two before the procedure, and eating or drinking dairy products is avoided the night before the start of the procedure. The procedure takes about 45 minutes and does not require sedation. Moreover, the colon and rectum needs to be cleansed the night before the test by laxative intake, and/or use of enemas the morning of the exam. At testing, a small flexible tube is inserted into the rectum, and barium sulfate liquid is pumped into it in order to partially fill and opens the colon. Air is then pumped into the colon through the same tube, which may lead to bloating, cramping and discomfort, in addition to an urge for a bowel movement. X-ray pictures of colon lining are taken. If polyps or other suspicious areas are observed, a colonoscopy may also be needed. The barium could cause constipation for a few days after the procedure, and there is a small risk due to inflating the colon with air, which could injure or puncture the colon, in addition to an exposure to a relatively small amount of radiation [4].
Colonoscopy, based upon the same principles as sigmoidoscopy, allows visualization of the entire colon. Although it is the “gold standard” for CRC screening for the 70 million people older than 50 years of age in the USA, it requires an unpleasant bowel preparation, the test itself could be uncomfortable, but sedation often helps, and some people could experience low blood pressure or changes in heart rhythm during the test due to the sedation, although these side effects are not serious. If polyps are removed or a biopsy is taken during the procedure, blood can be observed for a day or two after the test, and in rare cases when bleeding continues, it could require treatment [23]. The test costs about $10 billion per year and exceed the physician capacity to perform this procedure, requires cathartic preparation and sedating or anesthetizing the patient, and it has an increased risk of morbidity or mortality due to perforation of the GI [6,23]. Moreover, studies found the range of colonoscopy miss rates for right-sided colon cancer to be 4.0%, 12-13% for adenomatous polyps 6-9 mm, and 0-6% for polyps≥1 cm in diameter [4]. Clearly, a simple, inexpensive, noninvasive, sensitive and specific screening test is needed to identify people at risk for developing advanced adenomas (e.g., polyps≥1 cm with high grade dysplasia) or CRC who would benefit from a subsequent colonoscopy examination.
Virtual colonoscopy (CT colonography) is an advanced type of computed tomography (CT or CAT) scan of both the colon and rectum. It involves examination of a computer generated 3D presentation of the entire GI tract by reconstructing of either a computerized tomography (CT) or a magnetic resonance imaging. This test does not require sedation, but it requires bowel preparation and the use of a tube placed in the rectum --as in barium enema-- to fill the colon with air, and also the drinking of a contrast solution before the test in order to tag any remaining stool in the colon or the rectum. The procedure takes about 10 minutes, and it is especially useful for people who do not want to take the more invasive colonoscopy test. This method detects lesions based on their site, rather than their histology, and is thus unable to distinguish benign adenoma from an invasive carcinoma. It was shown in a meta-analysis of 33 studies involving 6,393 patients that this test has a low sensitivity for polyps (48% for polyps < 6mm, 70% for polyps 6-9 mm and 85% for polyps>9 mm). Moreover, the test is expensive, and requires the availability of experts, which could reduce patients’ compliance [25]. CT is still considered as an investigational alternative for asymptomatic, not at risk individuals, which also expose patients to a small amount of x-irradiation, and it can also miss the detection of small lesions [26].
In an effort to find a more pragmatic early biomarker noninvasive colon cancer detection methods, investigators have developed many in vitro tests such as epigenetic methylation marker changes in genes and chromosomal loci in fecal DNA [27], promoter DNA methylation in stool [28], mutated DNA markers found in neoplastic cells that are excreted in feces [29,30], or the minichromosomal maintenance proteins (MCMs) needed for DNA replication test [3], proteomics’-based approaches in stool or blood [31], and transcriptomic mRNA-based approaches in stool or blood [12], or a combination of both genetic, as well as epigenetic tests [32]. Molecular studies have shown the presence of mutations of K-ras in DNA from stool of patients, but its drawbacks include its expression by fewer than half of large adenomas and carcinomas. In addition, its expression in non-neoplastic tissue makes it less than an optimal molecular marker. Besides, mutations are only found in a portion of the tumor, making the test to be less sensitive [33].
Mutation of the adenomatous polyposis coli (APC) gene in stool of patients obtained by analysis of ductal DNA by PCR of APC gene templates and the detection of generated abnormal truncated polypeptides by in vitro transcription and translation of the PCR product has been demonstrated at early stages of the disease. However, the digital protein truncation test is not a reliable screening tool because it lacks specificity (i.e., 5 out of 28 controls were positive for FOBT, and another 6 showed rectal bleeding) [34]. Since CRCs exhibit genetic heterogeneity, a multitarget approach that employ mutations in K-ras, APC and p53; the microsatellite instability marker Bat-26; and “long” DNA representing DNA of nonapoptotic colonocytes characteristic of cancer cells exfoliated from neoplasms, but not normal apoptotic colonocytes, have been looked at and undergone clinical testing [35]. However, DNA alterations were detected in only 16 of 31 (51.6%) invasive cancer, 29 of 71 (40.8%) invasive cancer plus adenoma with high-grade dysplasia, and 76 of 418 (18.2%) in patients with advanced neoplasia (tubular adenoma≥1 cm in diameter), polyps with high grade dysplasia, or cancer [29]. Moreover, these tests are not cost-effective, as screening for multiple mutations is generally expensive [36].
Preliminary studies suggest that proteomics may distinguish normal state from adenoma. This approach has, however, not been evaluated as a noninvasive screening tool, and it is therefore considered investigational [37,38]. Currently, the markers most often elevated in advanced CRC are carcinoembryonic antigen (CEA) [39] and the carbohydrate antigen, which is also called cancer antigen (CA) 19-9 [40], but neither of these markers has been found to be a useful, or a reliable diagnostic screen for colorectal cancer.
Early detection would be greatly enhanced if accurate, practical and cost effective diagnostic biomarkers for CRC were available. However, despite the advances detailed above, tests now available neither detect colon cancer in all cases (i.e., have low sensitivity), nor are they highly specific. Furthermore, these tests are often costly, produce false-positive or false-negative results, molecules could be non-stable and easily fragment in vitro requiring excessive care and special handling techniques (mRNA molecules), and some methods entail discomfort/inconvenience to the patients, or could in rare cases result in mortality (e.g., colonoscopy) [21]; all are factors that could discourage patients’ enthusiasm and/or compliance. Current participation rates in CRC screening are less than 30% in both genders, compared to screening for breast and cervical cancer that have rates of 70 to 80%, respectively [41]. Participation could thus be enhanced by the use of molecular lab tests that are less uncomfortable, less expensive and offer greater accuracy (more sensitivity and specificity). However, larger clinical studies would be needed to corroborate initial test results.
On the other hand, our data and others [11,13,14,42-54], have shown that quantitative changes in the expression of few miRNA genes in stool or blood that are associated with colon cancer permit development of more sensitive and specific CRC molecular markers than those currently available on the market. In comparison to the commonly employed FOBT stool test, a noninvasive molecular and reliable test would particularly be more convenient as there would be no requirement for dietary restriction, or meticulous collection of samples, and thus a screening test would be acceptable to a broader segment of the population. Using stable molecules such as miRNAs that are not easily degradable when extracted from stool or blood and manipulated thereafter, a miRNA-approach for colon cancer is thus preferable to a xtranscriptomic mRNA-, mutation DNA-, epigenetic- or a proteomic-based test [11,42-56], particularly that we and others have shown that these stable, nondegradable miRNA molecules can be easily extracted from stool or from circulation in vitro using commercially available kits. Advantages and disadvantages of the in vivo and in vitro tests are presented in table 1.
| Table 1: Comparison of Tests Employed or Contemplated for Premalignant† & Malignant Colon Cancer Screening. | |||||||
| Test Specification |
FBOT†, Guaiac§ Immunolo● |
Methylated Gene■& chromosomal Loci | Promoter♦ Methylation | Mutated◘ DNA Markers |
Colonoscopy Examination▲ |
Proteomic Approach► |
MRNA▌ ,, miRNA▼ NGS₦ Approach Approach Approach |
| Noninvasive | Yes Yes | Yes | Yes | Yes | No | Yes | Yes Yes Yes |
| Sensitivity | 10.8%† 16.3%† | 7.5%■ | 31%† | 18.2% | 87% | 75% | >80% >90%¶, >95% |
| Specificity | 95% 94.5% | 82% | 95% | 94.4 | 100% | >95% | >95% >95%¶ >99% |
| Automation | No No | No | No | No | No | Yes | Yes Yes Yes |
| Cost | $15♪ $25♪ | $400♪ | $250♪ | $649♪ | $900♪ | $650♪ | $350♪ $200-400♪ $1000♪ |
| †,§,●FOBT: Is currently widely used because it is convenient and not costly, but it has low sensitivity and requires multiple sampling, which reduces compliance; from refs [7,9,15,18]. ■Methylated Gene: Only good for detection of advanced cancer, but not adenoma, based on vimentin gene, DY loci 5p21 and OC91199, thus it is not suitable for population screening; from ref [27]. ♦Promoter Methylation: Based on only one gene and is basically a specialized procedure for research purpose, thus not suited for population screening; from ref [28]. ◘Mutated DNA Markers: A DNA test that is marketed as Cologuard® and is distributed by Exact Sciences Corporation, Madison, WI includes qualitative analysis for KRAS mutations, abbreviated NDRG4 and BMP3 methylation, Badm, plus hemoglobin assay, for the presence of occult hemoglobin in human stool; from refs [29,30,32-35]. ▲Colonoscopy Examination: It is currently considered the Gold Standard for testing for colon cancer although it is invasive and could lead to certain risks in some individuals; from refs [7,18,21,32,35,36]. ►Proteomic Approach. Proteome ScantTM Technology uses LC-MS/MS to target proteomics quantitatively; however it needs specialized research equipment/reagents and is thus considered investigational; from refs [37,38,40]. ▌mRNA Approach: Targets changes in the fragile messenger RNA; therefore it requires special care in stool preparation, RNA extraction and storage, from refs [10,12,31]. ▼miRNA Approach: It is preferable than an mRNA approach because of the stability of the small miRNA molecules, and several miRNAs have good potential for population screening in either stool or blood. Based on our data for polyps ≥ 1 cm in diameter, villous or tubuvillous, or the presence of high grade dysplasia and carcinomas, from refs [48,52-56]. ₦NGS screening: It has potential, but needs further development before commercialization, from refs [112]. ♪Cost of a screening test depends on the number of miRNAs tested; estimates are based on citations, contacts with test developers, and our experience with clinical assay requirements and developments. |
|||||||
MicroRNAs as molecular markers for colon cancer screening in stool or blood
Stool testing has several advantages over other colon cancer screening media as it is truly noninvasive and requires no unpleasant cathartic preparation, formal health care visits, or time away from work or routine activities [3-6]. Unlike sigmoidoscopy, it reflects the full length of the colorectum and samples can be taken in a way that represents both the right, as well as the left side of the colon. It is also believed that colonocytes are released continuously and abundantly into the fecal stream [7,8], contrary to situation in blood--where it is released intermittently--as in FOBT [9], and transformed colonoctes produce more RNA than normal ones [10-14]; therefore, this natural enrichment phenomenon partially obviate for the need to use a laboratory technique to enrich for tumorigenic colonocytes. Furthermore, because testing can be performed on mail-in-specimens, geographic access to stool screening is unimpeded [2,16,32]. The American Cancer Society (ACS) (http://ww.cancer.org) has recognized that a promising diagnostic screen for CRC would be enhanced by employing a molecular-based stool testing.
It should be emphasized that although not all of the shed cells in stool are derived from a tumor, data published by us and others [11,13,14,44-56], have indicate that diagnostic miRNA gene expression profiles are associated with adequate number of exfoliated cancerous cells and enough transformed RNA is released in the stool, and also the availability of measurable amount of circulating. miRNA genes in blood (either cellular or extracellularly), which can be determined quantitatively by a sensitive technique such as PCR in spite of the presence of bacterial DNA, non-transformed RNA and other interfering substances. That quantification is feasible because of the high specificity of PCR primers that are employed in this method, which overcomes all of these stated obstacles; hence, the number of abnormally-shed colonocytes in stool, or total RNA presents in plasma or serum becomes unlimiting [11-14].
A test that employs miRNA in stool or blood could also result in a robust screen because of the durability of the miRNA molecules [11,13,14]. Moreover, an approach utilizing miRNA genes is more comprehensive and encompassing than a test that is based on the fragile messenger (m) RNA [12], for example, because it is based on mechanisms at a higher level of control. We believe that ultimately the final noninvasive test in stool or blood will include testing of several miRNA genes that show increased and decreased expression, and eventually a chip that contains a combination of these stable molecules will be produced to simplify testing, as has been developed for the testing of GMOs in foods [57].
Blood is a body fluid that can be obtained through a semi-invasive method (skin puncturing) that is commonly used in the laboratory testing, which makes it logical to employ on routine bases, and thus it would be attractive to technicians performing lab tests. However, working with blood for miRNA profiling present various challenges in purification and molecular characterization. For example, a naked miRNA molecule would degrade within seconds of vein puncture due to the presence of high levels of nucleases and other inhibitory components in blood, which can interfere with downstream enzymatic reactions, as for example, the common anticoagulant heparin that coamplify with RNA. Moreover, high-quality RNA preparations found in blood contain contaminants that inhibit a RT-qPCR reaction if too much sample is used in the RT preparation [58]. Therefore, it is recommended to use EDTA or citrate anticoagulated blood instead of heparin. Circulating miRNAs, however, have shown stability in several studies resulting from either the formation of complexes between circulating miRNAs and specific proteins [59-61], or the miRNAs are contained within protective circulating exosomes or macrovesicles [62]. Plasma is preferable to serum when quantifying miRNAs in blood, because its use minimizes variations caused by differences due to the lack of clotting factors [63].
For mature miRNAs testing, there are currently available commercial preparations that save time and provide the advantage of manufacturer’s established validation and QC standards. For example, a Qiagen buffer (miScript HiSpec Buffer®), Qiagen, Inc., Frederick, MD, USA, that inhibits the activity of the tailing reverse-transcription (RT) reaction on templates other than miRNA-sized templates provides for an exceptionally specific cDNA synthetic reaction that eliminates background from longer RNA species. To measure pre-miRNA, however, it would be essential to use another buffer (miScript HiFlex Buffer®) as the nonbiased reaction results in an increased background signal from cross reactivity with sequences from a total RNA preparation, which can be distinguished by performing a melt curve analysis when carrying out PCR analysis [64].
Small noncoding RNAs that exhibit little variation in different cell types (e.g., snoRNAs and snRNAs) are polyadenylated and are reverse transcribed (RT) in the same way as the small miRNAs and thereby could serve as controls for variability in sample loading and real-time RT-PCR efficiency. They are, however, not suited for data normalization in miRNA profiling experiments because they are not well expressed in serum and plasma samples. Therefore, normalization by a plate mean (i.e., mean CT value of all the miRNA targets on the plate), or using a commonly expressed miRNA targets (i.e., only the targets that are expressed in all samples are used to calculate the mean value) would be needed for a proper normalization of the amplification reaction [65].
An extraction protocol for miRNAs in blood can, however, be challenging. When setting up an extraction step, there are two options: either extract the miRNA molecules from cellular blood components, as whole blood is full of cells that can be obtained by differential centrifugation followed by isolating these cells, or from liquid plasma that contains circulating miRNAs. Attention, however, should be paid to heparin as this anticoagulant is known to be a strong inhibitor of polymerase in PCR reactions. There are several collection tubes that contain citrate as anticoagulant instead of heparin, as those made by Qiagen or Tempus can be used for the whole blood collection. If the aim is to isolate miRNAs from plasma, EDTA tubes can be used to collect blood and plasma isolated, then store at -80oC until ready for extracting the miRNAs, as these molecules are very stable under standardized laboratory extraction methods. Extraction can be carried out by modified Trizol method from Life Technologies, or miRNeasy reagent from Qiagen. Columns employed in extraction can be clogged and RNA may be lost and/or degraded; therefore, the integrity of total RNA needs to be checked on a standard agarose or acrylamide gels, or with an electrophoresis apparatus, like the Agilent Bioanalyzer. To check if RT-PCR method works, one should employ another source of RNA, as for example cells in culture. A RT- qPCR based screening, like hybrid based assays, however, does need validation. Both Life Technologies’ Taqman- and SYBR - based probes (like LNA Universal miRCURY RT microRNA PCR assay, made by Exiqon, Woburn, MA) have high specificity for short miRNAs and both methods showed similar efficiencies, without the need to design and validate home-made primers. MiRNA quantification by both methods, however, showed difference in variability that impact miRNA measurements, and therefore quantification is influenced by the choice of assay methodology. Thus, the method used for quantification must be considered when interpreting analyses of PCR results [66-69].
Our research team [11,13,14] and others [28,42-65,70-85] are in the opinion that a miRNA approach in tissue, cell lines, stool or plasma, could meet the criteria for test acceptability by laboratory staff carrying out these tests, as it is a non- or a minimally-invasive method, requites at the most 1 g of stool, or < 2 ml of blood (60% of which is plasma), does not need sampling on consecutive dates, can be sent by mail in cold packs, able to differentiate between normal subjects and colon adenomas/carcinomas, has high sensitivity and specificity for detecting advanced polyps, and can be automated, which makes it relatively inexpensive and more suited for early detection when compared to a test such as mutated DNA markers, especially since plasma is free from interfering clotting products, which are present in serum, miRNAs are stable in stool and plasma [11-14], and only 500 µl of plasma and 1 gram of stool, is required to perform the assay using commercially available kits [13,14]. The availability of powerful approaches for global miRNA characterization such as microarrays [86] and simple, universally applicable assays for quantification of miRNA expression such as qPCR [87] and statistical/bioinformatics methods for data analyses and interpretation [88-90], suggests that the validation pipeline that often encounters bottlenecks [15] will be more efficient in this assay. There is a pressing need for accelerating use of sensitive and stable molecular markers, such as miRNA molecules, in non- or minimally-invasive media such as stool and/or blood to improve the detection of CRC [91], particularly at an early tumor lymph node metastasis (TNM) disease stage (0-1) [92,93] while the cancer is still curable.
vThe discovery of small noncoding protein sequences, 17-27 nucleotides long RNAs, miRNAs, which regulate cell processes in ~ 30% of mammalian genes by imperfectly binding to the 3’ untranslated region (UTR) of target mRNAs resulting in prevention of protein accumulation by either transcription repression, or by induction of mRNA degradation [94,95], has opened new opportunities for a non-invasive test for early diagnosis of many cancers [53,66,70-81]. The latest miRBase release (v20, June 2013) [http://ww.mirbase.org] contains 24,521 21,264 miRNA loci from 206 species to produce 30,424 mature miRNA products [96]. Each miRNA generally targets hundreds of conserved mRNAs and several hundreds of nonconserved targets that operate in a complex regulatory network, and it is predicted that miRNAs together regulate thousands of human genes [49,54,56]. MiRNAs are transcribed as long primary precursor molecules (pri-miRNA) that are subsequently processed by the nuclear enzyme Drosha and other agents to the precursor intermediate miRNA (pre-miRNA), which in turn is processed in the cytoplasm by the protein Dicer to generate the mature single-stranded (ss) miRNA [97]. MiRNA functions have been shown to regulate development [98] and apoptosis [99] and specific miRNAs are critical in oncogenesis [51], effective in classifying solid [70-76] and liquid tumors [42,77-81], and serve as oncogenes or suppressor genes [100]. MiRNA genes are frequently located at fragile sites, as well as minimal regions of loss of heterozygosity, or amplification of common breakpoint regions, suggesting their involvement in carcinogenesis [101]. MiRNAs have great promise to serve as biomarkers for cancer diagnosis, prognosis and/or response to therapy [50,52,102]. Profiles of miRNA expression differ between normal tissues and tumor types, and evidence suggests that miRNA expression profiles clusters similar tumor types together more accurately than expression profiles of protein-coding mRNA genes [10,12,14,103].
Several of the miRNAs were shown by microarrays and RT-qPCR techniques in cell culture lines, CRC tissue, stool and blood to be related to colon cancer tumorigenesis [11,13,42,45-48,52,55,56,67,76,94] and ulcerative colitis (UC) [11]. A study indicated that a combination of mRNA and miRNA expression signatures represent a broader approach for improving biomolecular classification of CRC [103]. Another study employing microarrays and qPCR, in addition to an in situ hybridization test to assess differential expression in inflammatory bowel disease (IBD), showed aberrant expression of 11 miRNA in inflamed tissue and in HT-29 colon adenocarcinoma cells (3 showing significant decrease and 8 significant increase) [84]. Our work support the notion that quantitative changes in the expression of a few cell-free circulatory mature miRNA molecules in stool and plasma that are associated with colon cancer progression would provide for a more sensitive and specific biomarker approach than those tests that are currently available on the market [11,13,20,91].
As colon cancer-specific miRNAs are identified in stool colonocytes or blood plasma by microarrays- and qPCR-based approaches as presented in this review, the validation of novel miRNA/mRNA target pairs within the pathways of interest could lead to discovery of cellular functions collectively targeted by differentially expressed miRNAs [103]. For example, comparison of top 12 pathways affected by colon cancer and globally targeted by miRNAs overexpressed in CRC shows that coexpressed miRNAs collectively provide for a systemic compensatory response to the abnormal phenotypic changes in cancer cells by targeting a broad range of signaling pathways affected in that cancer [88].
Several algorithms such as: TargetScan [http://www.targetscan.org], DIANA-micro [http://www.diana.pcbi.upenn/edu], miRanda [1http://www.microrna.org], PicTar [http://pictar.bio.nyu.edu], EMBL [http://russell.embl-heidelberg.dr], EIMMo [http://www.mirz.unibas.ch], mirWIP [http://146.189.76.171/guery] and PITA Top [http://genic.weizmann.ac.il/ pubs/mir07/mir07 _data.html.1] have been used to correlate complementary 2-8 nucleotides seed sequences of mature miRNAs with target mRNA sequences in the 3’ UTR ends of in order to identify crucial control elements within a very complex regulatory system [85,87-90], that could be dysfunctional in CRC [104-107]. These programs differ in their requirement for base pairing of miRNA and target mRNA genes, and implement similar but not the same criteria when cross-species conservation is applied. Therefore, these different programs will invariably generate different sets of target genes for probably all miRNAs [91].
A study that examined global expression of 735 miRNAs in 315 samples of normal colonic mucosa, tubulovillus adenomas, adenocarcinomas proficient in DNA mismatch repair (pMMR), and defective in DNA mismatch repair (dMMR) representing sporadic and inherited CRC stages I-IV [108]. Results showed the following: a) six of the miRNAs that were differentially expressed in normal and polyps (miR-1, miR-9, miR-31, miR-99a, miR-135b and miR-137) were also differentially expressed with a similar magnitude in normal versus both the pMMR and dMMR tumors, b) all but one miRNA (miR-99a) demonstrated similar expression differences in normal versus carcinoma, suggesting a stepwise progression from normal colon to carcinoma, and that early tumor changes were important in both the pMMR- and dMMR-derived cancers, c) several of these miRNAs were linked to pathways identified for colon cancer, including APC/WNT signaling and cMYC, and d) four miRNAs (miR-31, miR-224, miR-552 and miR-592) showed significant expression differences (≥ 2 fold changes) between pMMR and dMMR tumors. The above data suggest the involvement of common biologic pathways in pMMR and dMMR tumors in spite of the presence of numerous molecular differences between them, including differences at the miRNA level [108].
Unlike screening for large numbers of mRNA genes, a modest number of miRNAs is used to differentiate cancer from normal, and unlike mRNA, miRNAs in stool and blood remain largely intact and stable for detection [11-14,19]. Therefore miRNAs are better molecules to use for developing a reliable noninvasive diagnostic screen for colon cancer, since we found out that: a) the presence of Escherichia coli does not hinder detection of miRNA by a sensitive technique such as qPCR, as the primers employed are selected to amplify human and not bacterial miRNA genes, and b) the miRNA expression patterns are the same in primary tumor, or in diseased tissue, as in stool and blood samples. The gold standard to which the miRNA test is to compared should be colonoscopy, which is obtained from patients’ medical records, as well as the cheaper immunohistological (IHC) FOBT screen, currently used in annual checkups, for comparison with miRNA results [18]. Although exosomal RNA will be missed [109], when using restricted extraction of total RNA from blood or stool, a parallel test could also be carried out on the small total RNA obtained from noninvasive stool or seminvasive blood samples, and the appropriate corrections for exsosomal loss can then be made after the tests are completed. A miRNA quantification workflow is presented in figure 1.
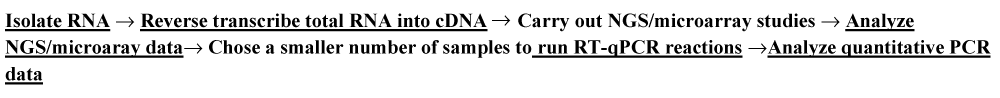
Figure 1: Experimental workflow for the quantification of miRNA molecules.
NGS, Microarray and RT-qPCR tests for quantitative detection of miRNAs in diversified samples
We have shown that we have been routinely and systematically able to extract a high quality total RNA containing miRNAs from a small number of laser capture microdissected (LCM) cells from tissue [110], colonocytes isolated from human stool [11,13,91] or circulating blood [14], using commercially-available kits (RNeasy isolation Kit®) from Qiagen, Valencia, CA, USA, followed by another kit from Qiagen “The “Sensiscript RT Kit”.
Next-generation sequencing (NSG) technologies
The 1977 chain-termination method of Sanger, commonly known as Sanger’s dideoxy sequencing [111], has been partly supplanted by other more cost effective next-generation sequencing technologies that provide higher throughput, but at the expense of read lengths. The Sanger method is based on DNA polymerase-dependent synthesis of a complementary DNA strand in the presence of 2’-deoxynucleotides (dNTPs) and 2’, 3’-dideoxynucleotides (ddNTPs) that serve as nonreversible synthesis terminators when ddNTPs are added to the growing oligonucleotide chains, resulting in truncated products of varying lengths, which can subsequently be separated by size on polyacrylamide gel electrophoresis. Advances in fluorescence detection have allowed for combining the four terminators into one reaction, using fluorescent dyes of different colors, one for each of the four ddNTP. Furthermore, the original slab gel electrophoresis was replaced by capillary gel electrophoresis, enabling better separation. Additionally, capillary electrophoresis was replaced by capillary arrays, allowing many in vivo amplified fragments samples cloned into bacterial hosts to be analyzed in parallel. Moreover, the development of linear polyacrlamide and polydimethylacrilamide allowed the reuse of capillaries in multiple electrophoretic runs, thereby increasing the sequencing efficiency. These and other advances of the sequencing technology have contributed to the relatively low error rate, long read length and robustness of modern Sanger sequencers. For example, the high throughput automated Sanger sequent instrument from Applied Biosystems (ABI 37730xl) has a 96 capillary array format that produces ≥ 900 PHRED 20 bp (a measure of the quality of identification of the nucleobases generated by sequencing) per read, for up to 96 kb, for a 3 h run [112].
The 454Roche instrument was the first next generation sequencer released to the market that circumvents the lengthy, labor intensive and error-prone technology by using in vitro DNA amplification known as emulsion PCR, where individual DNA fragment-carying streptavidin beads, obtained by the shearing the DNA and attaching the fragments to beads using adapters, which are captured into separate emulsion droplets that act as individual amplification reactors, producing ~107 clonal copies of a unique DNA template per bead. Each template-containing bead is then transferred into a well of a picotiter plate, which allows hundreds of thousands of clonally related templates of pyrosequencing reactions to be carried out in parallel, increasing sequencing output [113]. The sequence of DNA template is determined by a pyrogram, which corresponds to the correct order of chemiluminescently incorporated nucleotide as the signal intensity is proportional to the amount of pyrophosphate released. The pyrosequencing approach is prone to errors resulting from incorrectly estimating the length of homopolymeric sequence stretches (or indels). The Roche 454 platform, considered the most widely used next generation sequencing technology, is capable of generating 80-120 Mb of sequence in 200-300 bp reads in a 4h run [112].
The Illumina/Solexa approach achieves cloning-free DNA amplification by attaching a ssDNA fragment to a solid surface, known as a single molecule array, or free cell, and performing solid-phase bridge amplification of single molecule DNA templates in which one end of single DNA molecule is attached to a solid surface by an adapter; the molecule is subsequently bend over and hybridized to complementary adapters, creating a bridge, which serves as a template for the synthesis of complementary strands. Following the amplification, a flow cell containing more than 40 million clusters, each cluster composed of ~ 1000 clonal copies of a single tempelate molecule is produced. Templates are sequenced in massivley parallel manner using a DNA sequencing-by-synthesis approach that employs reversible terminators with removable fluorescent moieties and DNA polymerases capable of incorporating these terminators into growing oligonucleotide chains. The terminators are labeled with fluors of four different colors to distinguish among the different bases at the given sequence position, and the template sequence of each cluster is deduced by reading off the color at each successive nucleotide addition step. Although Illumina technology seems more effective at sequencing homopolymeric stretches than pyrosequencing, it produces shorter sequence reeds, and thus cannot resolve short sequence repeats. Moreover, substitution errors have been noted in this platform due to the use of modified DNA polymerases and reversible terminators. The 1G Illumina genome analyzer generates 35 bp reads per run in 2-3 days [115].
Massivley parallel sequencing (MPS) by hybridization-ligation supported in the oligonucleotide ligation and detection system SOLiD from Applied Biosystem is based on the polony sequencing technique [116]. Libraries begins with an emulsion PCR single-molecule amplification step, followed by transfer of the products onto a glass surface where sequencing occurs by sequential rounds of hybridizatrion and ligation with 16 dinucleotide combinations labeled by four different fluor dyes. Each position is probed twice and the identity of the nucleotide is determined by analyzing the color resulting from two successive ligation reactions. The two base encoding scheme allows the distinction between a sequencing error and a polymorphism (an error would be detected in only one reaction, whereas a polymorphism would be detected in both). The 1-3 GB SoLiD generates 35 bp reads per an 8 day run [114]. Table 2 illustrates available DNA sequencing technologies.
| Table 2: Available DNA sequencing technologies. | ||||
Technology* |
Approach |
Read length |
Bp/run |
Run time Company/web Reference |
| Automated Sanger sequencer 96 capillary array ABI3730xI | In vivo synthesis in the presence of dye terminator | From700 to 900 bp | 96 kb | AppliedBiosystems [111] 3h www.appliedbyosystems.com |
| 454/Roche FLX system | Pyrosequencing on solid support | 200-300 bp | 80-120 Mb | 4h Roche Applied Science [114] www.roche-applied-science.com |
| Illumina/Solexa | Sequencing by synthesis of single molecule arrays with reversible terminators | 30-40 bp | 1 Gb | 2-3h Illumina, Inc. http://www.illumina.com/ [115] |
| ABI/SOLiD | Massively parallel sequencing by ligation-hybridization | 35 bp | 1-3 Gb | 8d AppliedBiosystems [116] www.appliedbyosystems.com |
| * Helicos Genetic Analysis System Platform was the first conceived NGS implementationt using the principle of single molecule fluorescent sequencing for a standard 120 cycle 1100 field of view run, 12 to 20 x 106. It was marketed by Helicos Biosciences Corporation, Cambridge, MA, which filed Chapter 11 bankruptcy on November 15, 2015. | ||||
Microarray technologies
For microarray studies, we employed Affymetrix Gene Chip Micro 3.0 Array (Affymetrix, Inc, Santa Clara, CA, USA), which provides for 100% miRBase v17 coverage [http://ww.mirbase.org] by a one-color approach. The microarray contains 16,772 entries representing hairpin precursor, expressing 19,724 mature miRNA products in 153 species, and provides >3 log dynamic range, with 95% reproducibility and 85% transcript detection at 1.0 amol for a total RNA input of 100 ng.
Global microarray expression studies have shown similarity in expression between stool, plasma and tissue [117]. Microarray studies in stool samples obtained from fifteen individuals (three controls, and three each with TNM stage 0-1, stage 2, stage 3, and stage 4 colon cancer) showed 202 preferentially expressed miRNA genes that were either increased (141 miRNAs), or decreased (61 miRNAs) in expression [13].
A scatter plot comparing low dose microarray data to the control group presented in figure 2 shows a multigroup plot comparing miRNA -193a-3p to internal standard 18S rRNA in healthy normal control and the four TNM colon cancer groups (stages 0 to IV).
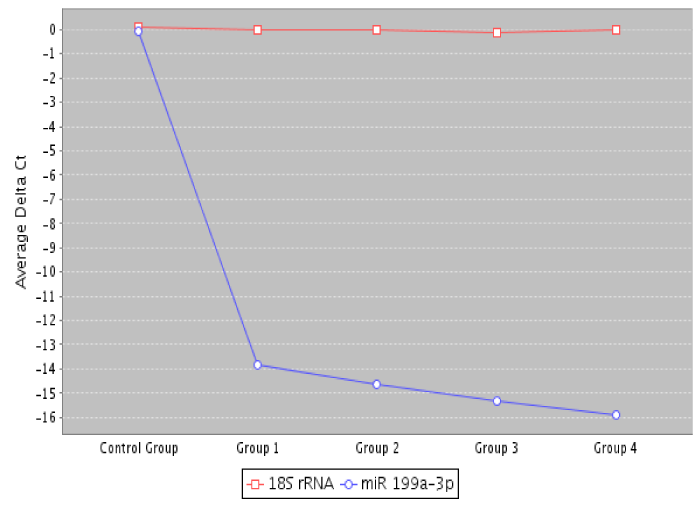
Figure 2: A multigroup plot comparing miRNA-199a-3p to normalization standard 18S rRNA in healthy controls and the four studied colon cancer groups, TNM stages 0 to 4.
To be able to screen several miRNA genes using the proposed PCR technology in a sequence-specific manner, in which a cDNA preparation can assay for a specific miRNA, we have employed in our work [11,13,14,91]. A sequence-specific stem-loop RT primers designed to anneal to the 3’-end of a mature miRNA, which result in better specificity and sensitivity compared to conventional linear ones [118]. This step was followed by a SYBR Green®-based real-time qPCR analysis in which a forward primer specific to the 5’-end of the miRNA, a universal reverse primer specific for the stem-loop RT primer sequence, and a 5’-nuclease hydrolysis probe-TaqMan minor grove binding (MGB) probe --matching part of the miRNA sequence and part of the RT primer sequence-- was employed in our Lab, using a standard TaqManPCR kit from Applied Biosystems on a Roche’s LightCycler (LC) 480 instrument, which employed the E-method [119], to calculate the relative expression of miRNA\ genes in modified RT-qPCR studies. It should be emphasized that the Roche’s LC-480 PCR instrument [120], employs a non-user-influenced method for high throughput measurements, using second derivative calculations and double corrections [121]. One correction utilizes the expression levels of a housekeeping gene of an experiment as an internal standard, which results in reduced error due to sample preparation and handling, and the second correction uses reference expression level of the same housekeeping gene for the analyzed expression in colonocytes or plasma, which avoids the variation of the results due to the variability of the housekeeping gene in each sample, especially in experiments that employ different treatments [122].
We conducted a stem-loop RT-TaqMan® minor groove binding (MGB) probes, followed by a modified qPCR expression assay on 20 selected mature miRNAs in stool, [13] and on 15 mature miRNAs in blood [14], that involved amplification of the gene of interest (target) and a second control sequence (reference) also called an external standard, which amplified with equal efficacy as the target gene, in the same capillary, a procedure known as “multiplex PCR”. Quantification of the target was made by comparison of the intensity of the products. A suitable reference gene has been the housekeeping pseudogene-free 18S ribosomal (r)RNA gene that was used as a normalization standard because of the absence of pseudogenes and the weak variation in its expression [123]. This selection has obviated the need to use normalization strategies such as plate mean (a mean CT value of all miRNA targets on the plate), a panel of invariant miRNAs [44], or commonly expressed miRNA targets [64]. A software to find a normalizer such as NormFinder [www.mld.dk/publicationnormfinder.htm], which is run as a template within Microsoft Excel® can also be used. For a more focused approach employing PCR on selected number of miRNA genes, we used miRNA stem-loop RT primers [118], for specific miRNA species to be tested, to make a copy of ss-DNA [11,13,14], for real-time PCR expression measurements.
Our RT-MGB PCR results in stool taken from 60 healthy controls and various stages of colon cancer patients are tabulated in table 3 and represented graphically by a scatter plot in figure 3. There has been no need to use receiver operating curves (ROCs) because the difference in miRNA expression between healthy and cancer patients, and among stages of cancer was large and informative. Data show that expression of 12 miRNAs (miR-7, miR-17, miR-20a, miR-21, miR-92a, miR-96, miR-106a, miR-134, miR-183, miR-196a, miR-199a-3p and miR214) had increased in stool of patients with colon cancer, and that later TNM stages exhibited a greater increased than did adenomas (Table 3). On the other hand, expression of eight miRNAs (miR-9, miR-29b, miR-127-5p, miR-138, miR-143, miR-146a, miR-222 and miR-938) was decreased in stool of patients with colon cancer that became more pronounced from early to later TNM stages (stages I to IV) [13]. A volcano plot depiction of quantification of mature miRNA by a stem-loop, TaqMan® MGB probes qPCR miRNA expression analysis of human stool for TNM group I using a Qiagen Corporation program [121], for colon cancer TNM stages 0-I is presented in figure 4.
| Table 3: Stem-loop RT, TaqManÒ MGB probes qPCR miRNA expression in stool from normal individuals & colon cancer patients. |
|||||||||||||||||||||
| Stool Sample |
18S rRNA stnd |
miR 199a-3p CP |
miR 196a CPa |
miR 183 CPa |
miR 96 CPa |
miR 7 CPa |
miR 214 CPa |
miR 21 CPa |
miR 20a CPa |
miR 92a CPa |
miR 134- CPa |
miR 17 CPa |
miR 106a CPa |
miR 143 CPa |
miR 146a CPa |
miR 9 CPa |
miR 222 CPa |
miR 138 CPa |
miR 127-5p CPa |
miR 29b CPa |
miR 938 CPa |
| NegCtb | 25.87 | 25.68 | 26.26 | 26.04 | 26.04 | 26.14 | 25.88 | 26.44 | 25.90 | 25.78 | 26.04 | 25.82 | 26.12 | 25.66 | 26.08 | 25.92 | 26.42 | 25.98 | 26.10 | 25.78 | 26.04 |
| N 1 | 25.98 | 26.04 | 25.56 | 26.91 | 26.06 | 26.27 | 26.32 | 25.66 | 26.52 | 26.14 | 25.90 | 26.15 | 25.77 | 25.68 | 25.65 | 25.80 | 25.78 | 26.02 | 25.98 | 26.06 | 25.36 |
| N 2 | 26.34 | 25.90 | 25.46 | 25.74 | 25.82 | 25.64 | 26.46 | 26.10 | 26.32 | 25.92 | 25.68 | 26.16 | 25.98 | 26.64 | 25.88 | 26.12 | 25.68 | 25.98 | 25.78 | 26.04 | 25.66 |
| N 3 | 26.46 | 26.04 | 25.84 | 26.22 | 26.34 | 26.18 | 26.44 | 26.52 | 26.22 | 26.16 | 25.98 | 25.88 | 26.10 | 26.14 | 25.88 | 25.98 | 26.12 | 26.06 | 26.08 | 25.68 | 26.04 |
| N 4 | 25.88 | 25.64 | 25.68 | 25.11 | 25.96 | 25.98 | 25.62 | 25.84 | 25.92 | 25.96 | 26.08 | 26.16 | 25.96 | 26.02 | 26.16 | 25.80 | 25.86 | 25.96 | 25.78 | 25.56 | 25.86 |
| N 5 | 26.78 | 25.69 | 26.24 | 25.78 | 25.94 | 25.86 | 25.48 | 25.86 | 25.94 | 26.22 | 25.88 | 25.92 | 26.12 | 25.98 | 26.12 | 25.88 | 26.16 | 25.78 | 26.12 | 25-98 | 26.04 |
| N 6 | 25.68 | 25.88 | 25.98 | 26.02 | 26.18 | 26.10 | 25.88 | 25.78 | 25.86 | 26.78 | 26.04 | 26.08 | 25.68 | 26.06 | 25.98 | 26.02 | 25.88 | 26.16 | 25.86 | 26.12 | 25.68 |
| N 7 | 25.66 | 26.02 | 26.10 | 25.98 | 25.84 | 25.88 | 26.14 | 25.68 | 26.06 | 25.68 | 25.98 | 25.78 | 26.10 | 25.66 | 26.08 | 25.76 | 26.06 | 25.68 | 25.98 | 25.86 | 25.76 |
| N 8 | 26.04 | 25.78 | 25.88 | 25.76 | 26.08 | 26.18 | 25.68 | 26.14 | 25.84 | 26.06 | 26.12 | 25.94 | 25.68 | 26.10 | 25.84 | 26.02 | 25.78 | 26.08 | 25.76 | 26.06 | 26.14 |
| N 9 | 25.68 | 26.04 | 26.08 | 25.88 | 25.92 | 25.86 | 26.06 | 25.76 | 26.10 | 25.66 | 25.82 | 26.06 | 26.02 | 25.82 | 26.08 | 25.78 | 26.14 | 25.86 | 26.02 | 25.98 | 25.80 |
| 10 | 26.12 | 25.80 | 25.92 | 26.06 | 25.74 | 26.08 | 25.90 | 25.84 | 25.94 | 26.08 | 26.04 | 25.90 | 25.80 | 26.06 | 25.72 | 26.06 | 25.96 | 25.88 | 25.90 | 26.12 | 26.06 |
| 11 | 25.78 | 26.10 | 26.04 | 25.88 | 26.08 | 25.84 | 26.02 | 26.06 | 25.72 | 25.90 | 25.68 | 26.08 | 26.14 | 25.94 | 26.06 | 25.88 | 26.16 | 26.06 | 26.10 | 25.90 | 25.80 |
| 12 | 26.02 | 25.96 | 25.78 | 26.08 | 25.96 | 26.12 | 25.78 | 25.88 | 26.06 | 26.08 | 26.12 | 25.76 | 25.96 | 26.08 | 25.90 | 26.08 | 25.76 | 25.88 | 25.96 | 26.04 | 26.12 |
| 13 | 25.68 | 26.02 | 26.10 | 25.82 | 26.06 | 25.76 | 26.10 | 26.06 | 25.90 | 25.76 | 25.96 | 26.06 | 26.10 | 25.84 | 26.16 | 25.76 | 26.14 | 26.08 | 26.08 | 25.96 | 25.88 |
| 14 | 26.02 | 25.96 | 25.76 | 26.00 | 25.96 | 26.08 | 25.80 | 25.96 | 26.06 | 25.80 | 26.08 | 25.76 | 25.74 | 26.06 | 25.88 | 26.16 | 25.92 | 25.88 | 25.90 | 26.06 | 26.16 |
| 15 | 25.86 | 26.08 | 26.14 | 25.98 | 26.02 | 25.80 | 26.12 | 25.98 | 25.86 | 26.02 | 25.90 | 26.08 | 26.12 | 25.88 | 26.04 | 25.78 | 26.06 | 26.10 | 25.86 | 25.90 | 25.84 |
| 16 | 26.00 | 25.92 | 25.86 | 26.08 | 25.96 | 26.08 | 25.90 | 26.16 | 26.08 | 25.78 | 26.06 | 25.78 | 25.66 | 26.12 | 25.70 | 26.06 | 25.96 | 25.80 | 26.02 | 26.12 | 26.02 |
| 17 | 25.70 | 26.04 | 26.08 | 25.96 | 26.14 | 25.78 | 26.10 | 25.84 | 25.90 | 26.04 | 25.96 | 26.10 | 26.06 | 25.94 | 26.16 | 25.86 | 26.06 | 26.02 | 25.92 | 25.76 | 25.92 |
| 18 | 26.06 | 25.76 | 25.94 | 26.06 | 25.82 | 26.14 | 25.88 | 26.06 | 26.02 | 25.86 | 26.12 | 25.86 | 25.96 | 25.78 | 25.84 | 26.06 | 25.96 | 25.86 | 26.10 | 26.04 | 26.00 |
| 19 | 25.86 | 26.06 | 26.16 | 25.86 | 26.08 | 25.94 | 26.02 | 25.82 | 25.78 | 26.08 | 25.90 | 26.08 | 26.12 | 25.98 | 26.10 | 25.80 | 26.06 | 26.14 | 25.76 | 25.86 | 25.94 |
| 20 | 26.04 | 25.96 | 25.84 | 26.06 | 25.86 | 26.08 | 25.86 | 26.04 | 25.92 | 25.88 | 26.08 | 25.86 | 25.96 | 26.12 | 25.78 | 26.04 | 25.76 | 25.86 | 26.02 | 26.10 | 25.84 |
| S0-11 | 26.04 | 12.08 | 13.066 | 14.04 | 15.12 | 16.06 | 17.02 | 18.04 | 19.08 | 20.16 | 21.20 | 22.28 | 23.62 | 35.50 | 34.44 | 33.40 | 32.22 | 31.16 | 30.12 | 29.10 | 28.08 |
| S0-12 | 25.90 | 12.10 | 13.04 | 14.08 | 15.24 | 16.18 | 17.14 | 18.10 | 19.16 | 20.18 | 21.18 | 22.24 | 23.50 | 35.56 | 34.36 | 33.46 | 32.28 | 31.20 | 30.18 | 29.16 | 28.12 |
| S0-13 | 26.06 | 12.12 | 13.12 | 14.20 | 15.16 | 16.12 | 17.12 | 18.16 | 19.08 | 20.14 | 21.16 | 22.18 | 23.54 | 35.60 | 34.40 | 33.48 | 32.26 | 31.24 | 30.22 | 29.18 | 28.22 |
| S0-14 | 25.96 | 12.14 | 13.16 | 14.26 | 15.26 | 16.16 | 17.10 | 18.20 | 19.22 | 20.16 | 21.22 | 22.22 | 23.60 | 35.54 | 34.38 | 33.44 | 32.20 | 31.18 | 30.26 | 29.20 | 28.26 |
| S0-15 | 26.08 | 12.16 | 13.10 | 14.30 | 15.24 | 16.26 | 17.06 | 18.18 | 19.21 | 20.18 | 21.20 | 22.20 | 23.58 | 35.58 | 34.42 | 33.42 | 32.24 | 31.22 | 30.24 | 29.14 | 28.10 |
| S0-16 | 25.90 | 12.12 | 13.08 | 14.16 | 15.14 | 16.24 | 17.16 | 18.14 | 19.24 | 20.22 | 21.24 | 22.14 | 23.52 | 35.58 | 34.40 | 33.52 | 32.30 32. |
31.26 | 30.28 | 29.16 | 28.14 |
| S0-17 | 25.86 | 12.16 | 13.02 | 14.20 | 15.28 | 16.14 | 17.08 | 18.12 | 19.16 | 20.24 | 21.26 | 22.16 | 23.44 | 35.62 | 34.50 | 33.54 | 32.36 | 31.28 | 30.14 | 29.22 | 28.18 |
| S0-18 | 25.92 | 12.18 | 13.16 | 14.24 | 15.32 | 16.18 | 17.18 | 18.16 | 19.18 | 20.20 | 21.28 | 22.26 | 23.38 | 35.66 | 34.46 | 33.46 | 32.38 | 31.30 | 30.16 | 29.12 | 28.16 |
| S0-19 | 25.86 | 12.06 | 13.14 | 14.28 | 15.22 | 16.20 | 17.22 | 18.20 | 19.28 | 20.22 | 21.30 | 22.32 | 23.36 | 35.34 | 34.48 | 33.48 | 32.34 | 31.34 | 30.20 | 29.24 | 28.20 |
| S0-110 | 26.02 | 12.22 | 13.18 | 14.20 | 15.36 | 16.42 | 17.20 | 18.22 | 19.30 | 20.28 | 21.36 | 22.36 | 23.46 | 35.36 | 34.44 | 33.50 | 32.30 | 31.32 | 30.28 | 29.26 | 28.24 |
| S0-111 | 25.94 | 12.16 | 13.08 | 14.18 | 15.24 | 16.38 | 17.24 | 18.10 | 19.24 | 20.26 | 21.34 | 22.34 | 23.48 | 35.42 | 34.52 | 33.38 | 32.32 | 31.36 | 30.32 | 29.30 | 28.28 |
| S0-112 | 25.88 | 12.20 | 13.10 | 14.32 | 15.30 | 16.34 | 17.18 | 18.16 | 19.28 | 20.18 | 21.24 | 22.38 | 23.50 | 35.46 | 34.56 | 33.34 | 32.42 | 31.44 | 30.36 | 29.28 | 28.22 |
| S0-113 | 26.08 | 12.04 | 13.14 | 14.30 | 15.38 | 16.44 | 17.22 | 18.12 | 19.26 | 20.20 | 21.28 | 22.26 | 23.54 | 35.48 | 34.54 | 33.32 | 32.48 | 31.40 | 30.34 | 29.34 | 28.30 |
| S0-114 | 25.78 | 12.14 | 13.20 | 14.28 | 15.30 | 16.38 | 17.26 | 18.14 | 19.16 | 20.22 | 21.26 | 22.30 | 23.46 | 35.52 | 34.46 | 33.36 | 32.46 | 31.42 | 30.30 | 29.36 | 28.36 |
| S0-115 | 25.92 | 12.12 | 13.18 | 14.36 | 15.18 | 16.26 | 17.22 | 18.18 | 19.24 | 20.18 | 21.32 | 22.32 | 23.38 | 35.58 | 34.36 | 33.40 | 32.44 | 31.48 | 30.38 | 29.38 | 28.44 |
| S0-116 | 26.04 | 12.08 | 13.12 | 14.40 | 15.32 | 16.24 | 17.26 | 18.24 | 19.28 | 20.14 | 21.34 | 22.40 | 23.40 | 35.60 | 34.50 | 33.42 | 32.40 | 31.50 | 30.42 | 29.40 | 28.32 |
| S0-117 | 25.96 | 12.10 | 13.14 | 14.22 | 15.26 | 16.36 | 17.32 | 18.26 | 19.22 | 20.26 | 21.36 | 22.38 | 23.24 | 35.44 | 34.52 | 33.44 | 32.50 | 31.36 | 30.40 | 29.32 | 28.42 |
| S0-118 | 26.04 | 12.14 | 13.08 | 14.34 | 15.14 | 16.24 | 17.38 | 18.16 | 19.14 | 20.28 | 21.30 | 22.40 | 23.28 | 35.50 | 34.60 | 33.46 | 32.56 | 31.38 | 30.36 | 29.44 | 28.40 |
| S0-119 | 25.88 | 12.06 | 13.10 | 14.38 | 15.18 | 16.12 | 17.30 | 18.24 | 19.26 | 20.24 | 21.38 | 22.36 | 23.42 | 35.56 | 34.38 | 33.32 | 32.52 | 31.44 | 30.34 | 29.42 | 28.38 |
| S0-20 | 26-06 | 12.10 | 13.06 | 14.44 | 15.28 | 16.10 | 17.26 | 18.22 | 19.24 | 20.12 | 21.20 | 22.24 | 23.50 | 35.64 | 34.32 | 33.36 | 32.54 | 31.46 | 30.44 | 29.46 | 28.36 |
| S2 1 | 26.12 | 11.22 | 12.20 | 13.34 | 14.24 | 15.66 | 16.62 | 17.40 | 18.08 | 19.14 | 20.16 | 21.26 | 22.56 | 36.12 | 35.12 | 34.16 | 33.20 | 32.18 | 31.22 | 30.18 | 29.16 |
| S2 2 | 25.94 | 11.34 | 12.16 | 13.44 | 14.30 | 15.54 | 16.44 | 17.32 | 18.24 | 19.18 | 20.22 | 21.28 | 22.50 | 36.16 | 35.18 | 34.20 | 33.26 | 32.26 | 31.26 | 30.24 | 29.22 |
| S2 3 | 25.88 | 11.28 | 12.08 | 13.22 | 14.26 | 15.36 | 16.52 | 17.38 | 18.36 | 19.24 | 20.18 | 21.34 | 22.54 | 36.20 | 35.26 | 34.28 | 33.28 | 32.28 | 31.28 | 30.22 | 29.26 |
| S2 4 | 25.96 | 11.30 | 12.24 | 13.28 | 14.38 | 15.42 | 16.56 | 17.40 | 18.34 | 19.26 | 20.24 | 21.36 | 22.48 | 36.26 | 35.28 | 34.36 | 33.24 | 32.34 | 31.24 | 30.20 | 29.18 |
| S2 5 | 26.08 | 11.16 | 12.26 | 13.34 | 14.28 | 15.34 | 16.38 | 17.36 | 18.38 | 19.20 | 20.26 | 21.32 | 22.44 | 36.24 | 35.30 | 34.38 | 33.22 | 32.20 | 31.20 | 30.30 | 29.20 |
| S2 6 | 25.90 | 11.32 | 12.22 | 13.42 | 14.34 | 15.20 | 16.46 | 17.34 | 18.40 | 19.22 | 20.12 | 21.30 | 22.32 | 36.30 | 35.16 | 34.44 | 33.30 | 32.22 | 31.30 | 30.22 | 29.24 |
| S2 7 | 25.88 | 11.42 | 12.26 | 13.46 | 14.32 | 15.28 | 16.36 | 17.28 | 18.36 | 19.28 | 20.20 | 21.20 | 22.42 | 36.36 | 35.20 | 34.42 | 33.36 | 32.30 | 31.26 | 30.26 | 29.28 |
| S28 | 25.86 | 11.36 | 12.40 | 13.44 | 14.36 | 15.32 | 16.24 | 17.32 | 18.38 | 19.30 | 20.30 | 21.18 | 22.38 | 36.38 | 35.24 | 34.48 | 33.32 | 32.40 | 31.30 | 30.24 | 29.30 |
| S2 9 | 26.04 | 11.38 | 12.28 | 13.38 | 14.28 | 15.38 | 16.36 | 17.28 | 18.40 | 19.22 | 20.32 | 21.22 | 22.42 | 36.28 | 35.30 | 34.46 | 33.34 | 32.24 | 31.34 | 30.28 | 29.34 |
| S210 | 25.98 | 11.28 | 12.34 | 13.32 | 14.30 | 15.30 | 16.42 | 17.34 | 18.26 | 19.28 | 20.16 | 21.24 | 22.32 | 36.32 | 35.32 | 34.40 | 33.38 | 32.36 | 31.36 | 30.32 | 29.32 |
| S31 | 25.78 | 10.80 | 11.20 | 12.44 | 13.36 | 14.28 | 15.32 | 16.24 | 17.30 | 18.24 | 19.18 | 20.22 | 21.36 | 37.40 | 36.38 | 35.28 | 34.20 | 33.18 | 32.22 | 31.26 | 30.18 |
| S32 | 26.02 | 10.84 | 11.36 | 12.36 | 13.44 | 14.34 | 15.42 | 16.30 | 17.36 | 18.30 | 19.26 | 20.26 | 21.38 | 37.46 | 36.42 | 35.34 | 34.26 | 33.24 | 32.26 | 31.28 | 30.22 |
| S33 | 25.96 | 10.72 | 11.42 | 12.30 | 13.40 | 14.36 | 15.36 | 16.32 | 17.32 | 18.26 | 19.20 | 20.28 | 21.34 | 37.48 | 36.48 | 35.30 | 34.28 | 33.26 | 32.24 | 31.30 | 30.20 |
| S34 | 25.88 | 10.78 | 11.28 | 12.34 | 13.48 | 14.42 | 15.38 | 16.28 | 17.38 | 18.28 | 19.22 | 20.32 | 21.30 | 37.42 | 36.44 | 35.32 | 34.24 | 33.28 | 32.20 | 31.24 | 30.24 |
| S35 | 26.06 | 10.66 | 11.34 | 12.40 | 13.52 | 14.44 | 15.44 | 16.36 | 17.40 | 18.30 | 19.24 | 20.20 | 21.40 | 37.44 | 36.40 | 35.36 | 34.22 | 33.32 | 32.28 | 31.26 | 30.26 |
| S41 | 26.08 | 10.02 | 10.60 | 11.38 | 12.42 | 13.50 | 14.56 | 15.48 | 16.56 | 17.44 | 18.38 | 19.14 | 20.22 | 38.28 | 37.32 | 36.28 | 35.32 | 34.28 | 33.28 | 32.32 | 31.30 |
| S42 | 25.94 | 10.08 | 10.42 | 11.32 | 12.36 | 13.42 | 14.48 | 15.56 | 16.62 | 17.48 | 18.32 | 19.24 | 20.26 | 38.34 | 37.34 | 36.30 | 35.26 | 34.24 | 33.26 | 32.36 | 31.26 |
| S43 | 25.84 | 10.10 | 10.56 | 11.28 | 12.30 | 13.38 | 14.40 | 15.50 | 16.48 | 17.52 | 18.40 | 19.22 | 20.28 | 38.36 | 37.36 | 36.36 | 35.36 | 34.22 | 33.34 | 32.34 | 31.24 |
| S44 | 25.90 | 10.04 | 10.60 | 11.12 | 12.22 | 13.30 | 14.34 | 15.52 | 16.54 | 17.46 | 18.42 | 19.26 | 20.20 | 38.30 | 37.42 | 36.42 | 35.34 | 34.26 | 33.30 | 32.38 | 31.28 |
| S45 | 26.02 | 10.06 | 10.52 | 11.18 | 12.32 | 13.42 | 14.48 | 15.44 | 16.50 | 17.42 | 18.30 | 19.28 | 20.24 | 38.32 | 37.46 | 36.38 | 35.28 | 34.30 | 33.32 | 32.30 | 31.22 |
| aComparative crossing point or (E-value): a value of test miRNA equals to the normalization standard indicates similar expression, a value lower than the standard indicates increased expression, and a value greater than the standard indicates decreased expression);bNo DNA added to reaction (negative control). All reactions were run in triplicates and then averaged. 18S rRNA is the normalization standard. The table shows expression values obtained from stool of 60 individuals that has been preserved in the stabilizer RNALater (Invitrogen, Carlsbad, CA): 20 non-cancerous controls (N 1 to N 20); 10 patients’ stool with adenomatous polyp ≥ 1cm (TNM stage 0-1); 10 patients’ stool with TNM stage 2 (S21-S310) colon cancer; 5 patients’ stool with TNM stage 3 colon cancer (S31-S35); and 5 patients’ stool with TNM stage 4 colon cancer (S41-S55). | |||||||||||||||||||||
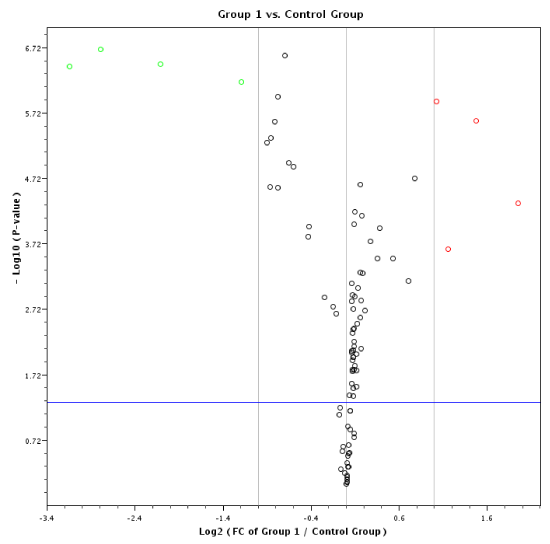
Figure 3: MicroRNA expression in stool samples taken from 60 healthy and colon cancer individuals. The stage of cancer is indicated by the bottom row of the panel. There were 20 normal healthy individuals, and 40 with colon cancer (TNM stages 0 to 4). Instances of high expression appear on the right and those with low expression on the left. Expression by stem-loop RT-minor grove binding qPCR was measured by the CP or the E-method on a Roche LightCycler® 480 PCR instrument. Scales were chosen so the minimum values line up on the “Min” mark labeled at top left of the panel. The same is true for the maximum values, which line up under the mark labeled “Max” at top right of the panel.
Figure 4: A volcano plot depiction of quantification of mature miRNA by a stem-loop, TaqMan® MGB probes qPCR miRNA expression analysis of human stool for TNM group I using a Qiagen Corporation program [115] for colon cancer TNM stages 0-I.
Of the selected 15 miRNAs that exhibited quantifiable preferential expression by qPCR in plasma, and have also been shown to be related to colon cancer carcinogenesis, nine of them (miR-7, miR-17-3p, miR-20a, miR-21, miR-92a, miR-96, miR-183, miR196a and miR-214) exhibited increased expression in plasma (and also in tissue) of patients with CRC, and later TNM carcinoma stages exhibited a more increased expression than did adenomas. On the other hand, six of the selected miRNAs (miR-124, miR-127-3p, miR-138, miR-143, miR-146a and miR-222) exhibited reduced expression in plasma (and also in tissue) of patients with colon cancer, with the reduction becoming more pronounced during progression from early to later TNM carcinoma stages [14].
Of the selected 15 miRNAs that exhibited quantifiable preferential expression by qPCR in plasma, and have also been shown to be related to colon cancer carcinogenesis, nine of them (miR-7, miR-17-3p, miR-20a, miR-21, miR-92a, miR-96, miR-183, miR196a and miR-214) exhibited increased expression in plasma (and also in tissue) of patients with CRC, and later TNM carcinoma stages exhibited a more increased expression than did adenomas. On the other hand, six of the selected miRNAs (miR-124, miR-127-3p, miR-138, miR-143, miR-146a and miR-222) exhibited reduced expression in plasma (and also in tissue) of patients with colon cancer, with the reduction becoming more pronounced during progression from early to later TNM carcinoma stages [14].
The stem-loop PCR stool data on 60 samples are tabulated in table 3 and presented graphically in figure 3 using a scatter plot, and also in figure 4 employing a volcano plot exhibits minimal variance within groups resulting in low p-values calculated using 2(-dCT) (SD of 0.015275 or 0.025166 is minimal, or raw CT values is only~0.03 for three replicates). The 95% CT for group 4 was between 134.39 and 135.63, an indication of a slight variation between groups. However, because the raw CT variations are low, even the slightest changes resulted in significant p-values; for example, miR-193a-5p was induced in different groups by between two to 134-fold (Table 4). It should be emphasized that there was been no need to use receiver operating characteristic (ROC) curves because the difference in miRNA expression between healthy individuals and patients with colon cancer, and among stages of cancer was large and informative.
| Table 4: Standard deviations (SDs) for some miRNAs in order of decreasing values. | ||||||||||
| miR-96 7.898177 |
miR-143 7.295028 |
miR-146a 6.593613 |
miR-214 6.550193 |
miR-21 6.356752 |
miR-9 6.042022 |
miR-7 5.793815 |
miR-92a 5.623533 |
miR-20a 5.450223 |
miR-134 5.288764 |
miR-938 5.204872 |
| miR-222 5.193460 |
miR-138 4.789436 |
miR-127-5p 4.139903 |
miR-29b 3.804948 |
miR-17 3.796239 |
miR-183 0.612726 |
miR-196a 0.531256 |
miR-199a-3p 0.379780 |
miR-106a 0.144222 |
18SrRNA 0.5513392 |
|
For example, the presented data can be compared to that which would be obtained from a group of students where half are 1st graders and the other half are high school students (although we have considered more groups, the idea can still be exemplified with just two groups). To separate these groups, we would use height as a measurement (in our experimental work we used gene expression). It turns out that the shortest high school student is a lot taller than the tallest 1st grader and all those above are high school students. Specificity, sensitivity and area under the curve are all 100%. When we use weight (in our work, a different expression) we get the same results: the lightest high school student is a lot heavier than the heaviest 1st grader. We can use other measures, such as shoe size or reading level, and again we get the same result.
Thus, our results are in general agreement with what has been reported in the literature for the expression of these miRNAs in tissue, blood, stool of colon cancer patients, and cells in culture [42-46,48,50,52,54-56,67,76,103]. This indicates that the choice of carefully selected set of miRNAs can distinguish between non-colons from colon cancer, and can even separate different TNM stages. A miRNA expression index similar to that developed for mRNA [124] or a complicate multivariate statistical analysis [125], was therefore not necessary in this case in order to reach conclusions from these data.
The initial number of miRNA genes (whether 15 or twenty) could be refined by validation studies to a much lower number (or even a single miRNA molecule) if the data pans out in a larger epidemiologically randomized study [126] that employs a prospective specimen collection retrospective blinded evaluation (PRoBE) design for randomized selection of control subjects and case patients from a consented cohort population, to avoid bias and to ensure that biomarker selection and outcome assessment will not influence each other, in order to have a statistical confidence in data outcome. The validated miRNAbiomarkers can then be placed on a chip to facilitate screening, as has been done for the testing of genetically modified organisms in food [57], to facilitate and automate studying miRNA expression./p>
It is necessary to clearly understand the normal, healthy functions of the human body, and their value ranges (e.g. with respect to age, sex, environment), in order to more thoroughly detect what is abnormal by studying human tissue/blood/stool from healthy donors and patients. Such studies need high quality samples from large numbers of subjects--in the hundreds to thousands-designed by an appropriate epidemiological method that employs a randomized unbiased PRoBE design of hundreds to thousands of control subjects and case patients from a consented cohort population [127].
Method for PCR quantification, normalization and quality control issues
The comparative cross point (CP) value (or E-method) [119] was employed, utilizing the LightCycler (LC) Quantification Software™, Version v4.0 [120], for Roche LC PCR instruments (Mannheim, Germany) for the semi-quantitative PCR analysis. The method employs standard curves in which the relative target concentrations is a function of the difference between crossing points (or cycle numbers) as calculated by the second derivative maximum [121], in which the Cycler’s software algorithm identifies the first turning point of the graph showing fluorescence versus cycle number to calculate the expression of miRNA genes automatically without user’s input, with a high sensitivity and specificity. A CP value corresponds to the cycle number at which each well has the same kinetic properties. The CP method corresponds to the 2,-ΔΔCT method [128], used by other PCR instruments, although the latter method produces reliable quantitative results only if the efficiency [E=10-1/slope] of the PCR assay for both target and reference genes are identical and equal to 2 (i.e., doubling of molecules in each amplification cycle); for example if well A1 has a CP value of 15 and well A2 has a CP value of 16, we deduce that there was twice as much of the gene of interest in well A1. A 10-fold difference is shown by a difference of ~ 3.3 CP value. It is not possible to compare these values between different primer pairs. The CP method compensates for difference in target and reference gene amplification efficiency either within an experiment, or between experiments.It is also essential to normalize the data to a “reference” housekeeping internal standard gene (e.g., endogenous reference genes RNU6 genes RNU6A and RNU6B, SNORD genes SNORD43, SNORD44, SNORD48, SNORA74A) or miRNA normalizers (e.g., miRNA 16, miRNA-191), or in some cases against several standards because the total input amount may vary from sample to sample when doing relative quantification. To ensure that miRNA quantification is not affected by the technical variablility that may be introduced at different analysis steps, synthetic nonhuman spike-in miRNA have been used to monitor RNA purification and RT efficiencies. The C. elegans cel-miR-39, cel-miR-54, the synthetic miRNAs Quanto ECI and Quanto EC2, and the simian virus gene SV40 have been used; these exogenous miRNA are usually added to samples before the RT step to avoid differences in template quality, or affect the efficency of the RT reaction, and can eliminate deviation of the results, making results reliable, but does not corrrect for sampling deviation or quality of tissues, body fluid or extracellular vesicle samples. It has been proposed that the best normalization strategy is the one that employs a combination of exogenous and endogenous control miRNAs because this compensates for differences in miRNA recovery and cDNA synthesis among samples [128]. Some studies used absolute data normalization and calculated miRNA expression using standard curves developed by synthetic miRNA and melting curves normalized per nanogram of the total input RNA for miRNA-221 and miRNA-18a in 40 pairs of CRC tissue and 595 stool samples, a technical detection limits of 2 copies for miRNA-221 resulted in a Cq value of 42, and a technical detection limit of 5 copies for miRNA-18a resulted in a Cq value of 47, which were all assigned a value of 0, similar samples with no amplification of miRNA-221 or miRNA-18a [129]. It should note, however, that values of Cq>40 are unreliable [128]. Absolute normalization method is thus considered to be reliable only for samples with good RNA quality [130].
To report “fold change” results, the LC software incorporates all those factors. The CP method can normalize for run-to-run differences, as those caused by variations in reagent chemistry. For such normalization, one of the relative standards must be designated a ”calibrator” for the target and for the reference genes, which can be any of our healthy control stool sample. These calibrator(s) can then be used repeatedly in subsequent runs to guarantee a common reference point, allowing for comparison of all experiments within the series. If necessary, the 2-ΔΔCT can be calculated by instrument’s software if samples are properly labeled; the 2-ΔΔCT calculations can also be set up manually. To determine fold change for a particular unknown cancer stool or blood sample that has a target gene CP value of 10, one needs three additional values: a) The reference gene CP value of that same unknown stool sample/cancer stool sample, b) the target gene CP for the calibrator sample/normal stool, and c) the reference gene CP for the calibrator sample/normal stool or blood [131].
In all PCR reactions, strict attention must be given to quality control (QC) procedures, and as the field has matured, guidelines on reporting qPCR data known as minimum information for publication of quantitative real-time PCR expression (MIQUE) has also been implemented by us [132], in order to ensure the uniformity, reproducibility and reliability of the PCR reaction and data integrity.
Statistical methods and bioinformatics analyses
In genomics work, it is important to have an understanding of statistics and bioinformatics to appreciate and make sense of generated data [133]. First, power analysis could be used for estimating sample size for a study [135]. Moreover, power analysis, as well as first and second order validation studies could be carried out to access the degree of separation and reproducibility of the data [135].
If the difference in miRNA gene expression between healthy and cancer patients and among the stages is found to be large and informative for multiple miRNA genes, suggesting that classification procedures could be based on values exceeding a threshold, then a sophisticated classification would not be needed to distinguish between the study data. However, if inconsistent differences on large samples are found, then predictive classification methods can be employed [13]. Programs supplied by Qiagen Corporation can be used free of charge to analyze, normalize and graph molecular data (http://pcrdataanalysis.sabiosciences/com).
The goal in predictive classification will be to assign cases to predefined classes based on information collected from the cases. In the simplest setting, the classes (i.e., tumors) are labeled cancerous and non-cancerous. Statistical analyses for predictive classification of the information collected (i.e., microarrays and qPCR on miRNA genes) attempt to approximate an optimal classifier. Classification can be linear, nonlinear, or nonparametric [133,135]. The miRNA expression data could be analyzed first with parametric statistics such as Student t-test or analysis of variance (ANOVA) if data distribution is random, or with nonparametric Kruskall-Wallis, Mann-Whitney and Fisher exact tests if distribution is not random [133,136]. If needed, complicated models as multivariate analysis and logistic discrimination [137,138] could also be employed.
False positive discovery rates (expected portion of incorrect assignment among the expected assignments) could also be assessed by statistical methods [139-141], as it could reflect on the effectiveness of the test, because of the need to do follow up tests on false positives. The number of optimal miRNA genes (whether 20 or less) to achieve an optimum gene panel for predicting carcinogenesis in stool will need to be established by statistical methods.
For the corrected index, cross-validation could be used to: protect against overfitting, address the difficulties with using the data to both fit and assess the fit of the model, and determine the number of samples needed for a cancer study, where the expected proportion of genes’ expression common to two independently randomly selected samples is estimated to be between 20% and 50% [142]. Efron and Tibshirani [143], suggested dividing the data into 10 equal parts and using one part to assess the model produced by the other nine; this is repeated for each of the 10 parts. Cross-validation provides a more realistic estimate of the misclassification rate. The area under the ROC curves, [in which sensitivity is plotted as a function of (1-specificity)], are used to generally describe the trade-off between sensitivity and specificity [144].
Principal component analysis (PCA) method [145], which is a multivariate dimension reduction technique, could also be used to simplify grouping of genes that show aberrant expression from those not showing expression, or a much reduced expression. In cases where several genes by themselves appear to offer distinct and clear separation between control or cancer cases in stool samples, a PMI may thus not be needed.
If the miRNA gene panel (or a derived PMI) is found to be better than existing screening methods, then all of the data generated can be used to assess the model so over-fitting is not a concern. The level of gene expression could be displayed in a database using parallel coordinate plots [146,147], produced by the lattice package in R (version 2.9.0, The R Foundation for Statistical Computing [http://cran.r-project.org], and S-plus software (Insightful Corporation, Seattle, WA). Other packages such as GESS (Gene Expression Statistical System) published by NCSS [http://www.ncss.com] could also be employed, as needed.
Bioinformatics analysis using the basic TargetScan algorithm for up-regulated and down regulated mRNAs genes has been employed. The program yielded 21 mRNA genes encoding different cell regulatory functions. The first 12 of these mRNAs were found with the DAVID program [148], to be active in the nucleus and related to transcriptional control of gene regulation. For down regulated miRNAs, the DAVID algorithm found the first four of these mRNAs to be clustered in cell cycle regulation categories [12].
Tumor heterogeneity due to mismatch DNA repair
To add another level of complexity to colon cancer, colon tumors have shown differential expression of miRNAs depending on their mismatch repair status. MiRNA expression in colon tumors has exhibited an epigenetic component, and altered expression due to mismatch repair may reflect a reversion to regulatory programs characteristic of undifferentiated proliferative developmental states [149].
MiRNAs also undergo epigenetic inactivation [150], and miRNA expression in CRC has been associated with MSI subgroups [151,152]. MiRNAs may regulate chromatin structure by regulating key histone modification; for example, cartilage-specific miR-140 targets histone deacetylase 4 in mice [153], and miRNAs may be involved in meiotic silencing of unsynapsed chromatin in mice [154]. In addition, DNA methylation enzymes DNMT1, 3a and 3b were predicted to be potential miRNA targets [155]. Moreover, a specific group of miRNAs (epi-miRNAs), miR-107, -124a, -127, directly target effectors of the epigenetic machinery such as DNMTs, histone deacetylases and polycomb repressive complex genes, and indirectly affect the expression of suppressor genes [156-158].
In addition to negatively regulating target mRNA, miRNAs are regulated by other factors. For example, c-myc activate transcription of miR-17-92 cluster that has a role in angiogenesis [159], and TFs NFI-A and C/EBPα compete for binding to miR-223 promoter decreasing and increasing miR-223 expression, respectively [160]. MiR-223 also participates in its own feedback, and favors the C/EBPα binding by repressing the NFI-A translation. Many of the miRNAs located in the introns of protein-coding genes are co-regulated with their host gene [161]. The challenge now is to identify those driver methylation changes that are thought to be critical for the process of tumor initiation, progression or metastasis, and distinguish these changes from methylation changes that are merely passenger events that accompany the transformation process but that have no effect per se on carcinogenesis.
Test performance characteristics (TPC) of the miRNA> approach
Cytological methods carried out on purified colonocytes employing Giemsa staining [162], as described for CRC, showed a sensitivity for detecting tumor cells in smears of 80%, which is slightly better than that reported earlier (i.e. about 78%) [163,164].
Numerical underpinning of the miRNAs as a function of total RNA was carried out on colonocytes isolated from stool [165], before any preservative was added to five healthy control samples, and five TNM stage IV colon cancer samples, extracting total RNA from them and determining the actual amount of total RNA per stool sample, and from the average CP values, taking into account that some exsosomal RNA will not be released from purified colonocytes into stool, and arbitrarily corrected for that effect [166]. It is evident from data shown in table 5 that an average CP value for stage IV colon carcinoma of 21.90 is invariably different from a CP value of 26.05 for healthy controls.
| Table 5: Numerical underpinning of miRNA markers as a function of total RNA. | ||||
| Patient # | Diagnosis | Total RNA (µg/g of stool) |
_CP* | |
| Sample | Group | |||
| 2 | Healthy control | 0.28 | 25.99 | |
| 5 | Healthy control | 0.30 | 27.21 | |
| 8 | Healthy control | 0.29 | 25.96 | 26.05 |
| 14 | Healthy control | 0.31 | 25.09 | |
| 17 | Healthy control | 0.32 | 26.01 | |
| 56 | Stage IV carcinoma | 0.31 | 20.18 | |
| 57 | Stage IV carcinoma | 0.32 | 22.03 | |
| 58 | Stage IV carcinoma | 0.31 | 22.43 | 21.90 |
| 59 | Stage IV carcinoma | 0.30 | 21.37 | |
| 60 | Stage IV carcinoma | 0.31 | 23.49 | |
| CP*: Crossing point value calculated by an algorithm in a Roche LC 480 polymerase chain reaction instrument. | ||||
Test performance characteristics (TPC) of the miRNA approach obtained by the CP values of the miRNA genes calculated from stool colonocyte samples of normal healthy individuals and patients with colon cancer were compared to the commonly used FOBT test and with colonoscopy results obtained from patients’ medical records in 60 subjects (20 control subjects and 40 colon cancer patients with various TNM stages). The data showed high correlation with colonoscopy results obtained from patients’ medical records for the controls and colon cancer patients studied.
Conclusions and Recommendations
The innovation of employing a miRNA approach for colon cancer screening lies in the exploratory use of an affordable, quantitative miRNA expression profiling of few of these molecules in noninvasive stool or semiinvasive blood samples, whose extracted fragile total RNA can been stabilized in the laboratories shortly after stool collection or blood drawing by commercially available kits so it does not ever fragment, followed by global miRNA expression, then quantitative standardized analytical real-time qPCR tests on fewer selected genes that are neither labor intensive, nor requires extensive sample preparation, in order to develop a panel of few novel miRNA genes for the diagnostic screening of early left and right sporadic colon cancer more economically, and with higher sensitivity and specificity than any other colon cancer screening test currently available on the market.
RT-qPCR has been the subject of considerable controversy. While the technique is considered the gold standard for quantifying gene expression in a cell, tissue or body fluid/excrement, there are so many variables involved that different labs could perform the same experiment and end up with different results. Moreover, although a study may produce a statistically significant result, it’s hard to know if that result is truly valid or if the data might have been skewed due to a technical error. Therefore, in 2009, a group of researchers published guidelines to help scientists publish data that are both accurate and reproducible. These guidelines are known as “The Minimum Information for Publication of Quantitative Real-Time PCR Experiments (MIQE)”. They address several key aspects of qPCR, including sample quality control, assay design, PCR efficiency, and normalization. A paper that attempted to identify a set of suitable, reliable reference genes for several different human cancer cell lines and to determine whether or not MIQE guidelines are followed, reported that in many of the studies important data are missing, as many publications do not report the efficiency of their reference genes or their qPCR data, and that only 30-40 percent of published studies that investigated reference genes actually followed the MIQE guidelines [132]. Moreover, as the newest incarnation of PCR, digital pCR or dPCR, is now being used by an increasing number of labs to provide for broader quantification, a new set of MIQE guidelines geared to the specific concerns of this brand-new version of PCR have recently been published [167].
It is noteworthy to point out that since the discovery of miRNA in 1993, investigators working in cancer research paid attention to these regulatory molecules and attempted to develop minimally-invasive markers to diagnose this disease. Although methods that employ PCR in stool and blood samples are currently in the forefront of the quantitative methods to develop reliable screening markers, a chip that contain a combination of these genes could be produced to simplify testing, as has been accomplished in testing of genetically modified organisms in foods [57].
MiRNAs are interesting biomarkers that are stable, amplifiable, and functionally important, have ample information content, play a significant role in gene regulation, and the expression profiles of the miRNAs annotated in miRBase release 20, June 2013 was 24, 5211 loci in 206 species using small RNA deep sequencing 800 validated molecules allows for distinguishing malignant and non-malignant tissue, as well as distinguishing different tumor entities [96]. Most circulating miRNAs are associated with Argonaute2, which is part of the RISC silencing complex. But whether these circulating miRNAs come from normal tissue or tumor tissue and how they are released into body fluids-through cell death or some other process-are mostly unanswered questions. In healthy tissue, evidence indicates that cells release miRNAs, both in vesicles and in protein complexes, which can then act as intercellular signaling molecules. When taken up by a recipient cell, miRNAs could modulate their gene expression. In tumor tissue cells they promote a microenvironment that helps the tumor survive, giving tumors a selective advantage. However, it is not known what is the balance between passive release by various ways, and release that is programmed within the cell, as for example, immune cells. Many circulating miRNAs linked to solid tumors are also expressed in blood cells. The source of miRNAs is not important, provided they are validated as markers. What has been a challenge is to establish standardized protocols for extracting and quantifying circulating miRNAs, as the technology keeps developing and improving; however, it is expected that in 5 to 10 years, we’ll have worked out the best way to quantitate miRNAs in blood and other body fluids.
Because results for many tumor markers have not been adequately reported, this anomaly has led to difficulty in interpreting research data and inability to compare published work from different sources, guidelines for carrying out tumor marker studies in a transparent fashion and for adequately reporting research findings have been jointly published by the USA National Cancer Institute and the European Organization for Research and Treatment of Cancer (NCI-EORTC) [168], so that researchers could have confidence in outcome and could repeat these data using the published methods.
It is envisioned that eventually a microfluidic device of an implantable biosensor platform that is simple in design, durable in performance and easy to use will be produced, whereby an individual takes noninvasive stool or semi-invasive blood samples at home and inserts them into it for assay of colon cancer disease markers. Identification of early stage disease biomarkers combined with a realistic awareness of self and sustained discipline for good and improved health would allow the individual to take preventative actions quickly, which will help prevent the spread of this cancer.
The following recommendations are considered important and represent a summary of how we envision miRNAs to influence colon cancer development and progression:
1. It is necessary to thoroughly understand the normal, healthy functions of the human body, and their value ranges (e.g. with respect to age, sex), in order to more rapidly detect what is abnormal.by studying human tissue/blood/ stool from healthy donors and patients. Such studies need high quality samples from large numbers of subjects (in the hundreds to thousands) selected by an appropriate epidemiological design to facilitate reaching meaningful conclusions.
2. When carrying out biological studies, it is essential to select the number of subjects by an epidemiologically-acceptable approach, and to have an adequate number of samples (in the hundreds to thousands) to be able to carry out a thoughtful analyses, and to be able to reach meaningful conclusions.
3. In its application as a screening approach, global miRNA profiling by a high throughput omic method such as next generation sequencing (NGS) and microarrays, followed by real-time qPCR, as well as digital PCR (dPCR) should be looked at as an expedition into the terra incognita of molecular diagnosis to identify novel genes, mechanisms and/or pathways in which a stimuli, whether genetic or environmental, exerts a change on the physiology of the cell.
4. MiRNA profiling is limited by available cells, which could be obtained by noninvasive methods, genetic heterogeneity of the tested population, and environmental factors such as diverse life styles and nutritional habitats.
5. MiRNA quantification can be influenced by the choice of methodology, which must be considered when interpreting the miRNA analysis results.
6. Because array data often underestimate the magnitude of change in miRNA level, it would be essential to use an independent confirmatory method such RT-qPCR, or Northern blotting, to check the magnitude of miRNA level of the identified target gene(s), as the magnitude of the change in the miRNA level depends on a variety of parameters, particularly the employed normalization method.
7. It is essential to mormalize PCR data to a reference standard(s) using either endogenous satandards, or exogenous miRNA, but preferably a combination of both.
8. To avoid errors due to exosomal RNA loss using restricted extraction of total RNA from stool or blood, a parallel test should also be carried out on total RNA obtained from stool or plasma samples, and appropriate corrections for exsosomal loss need to be made.
9. Although it is mainly used now as a basic science tool, global miRNA gene expression is moving from laboratories to large-scale clinical trials as a diagnostic tool to describe a pathophysiologic condition, or even allow clinical states to be determined in diseases such as cancer.
10. MIQE guidelines, which address several key aspects of qPCR, including sample quality control, assay design, PCR efficiency, and normalization were published in 2009 to help scientists publish data that are both accurate and reproducible, which have been followed recently by similar guidelines for digital PCR.
11. Aquiring a signature of several miRNAs will provide useful information for the clinicians to make decision on personalized management of the disease, instead of a single miRNA marker.
12. Although our results show that several miRNA genes can be used to discriminate noninvasively healthy individuals from patients with colon cancer, it would, however, be necessary to conduct a prospective randomized validation study using the methods that we have outlined herein, but on a much larger number of individuals to have a statistical confidence in data outcome.
13. Effort is needed to identify driver methylation changes believed to be critical to the process of tumor initiation, progression or metastasis, and distinguish these from methylated changes that are passenger events, accompanying the transformation process but have no effect per se on carcinogenesis.
14. Guidelines for carrying out tumor marker studies in a transparent fashion and for adequately reporting research findings have been jointly published by the US National cancer institute and the European Organization for Research and treatment of cancer (NCI-EORTC).
Acknowledgments
We express my gratitude to Drs. Paul Vos and Clark Jeffries for insight in biostatistics and bioinformatics, respectively. Informed written concent was taken from all study participants. There has been no conflict of interest in carrying out this research, or in the preparation of the article for publication.This work has been supported by NIH Grant 1R43-CA144823-A1-01 from the Department of Health and Human Services, National Cancer Institute, National Institutes of Health, Bethesda, Maryland, USA.; the State of North Carolina SBIR/STTR Matching Funds Program, Grant # G30433001211SBIR from Office of Science and technology, Raleigh, NC, U.S.A.; and additional operating funds from GEM Tox Labs, Institute for Research in Biotechnology, Greenville, NC, USA.
References
- Peterson NB, Murff HJ, Ness RM, Dittus RS. Colorectal cancer screening among men and women in the United States. J Womens Health. 2007; 16: 57-65. Ref.: https://goo.gl/3EDoae
- Mandel JS. Screening for colorectal cancer. Gastrointestinal Clin N Ame. 2008; 37: 97-115. Ref.: https://goo.gl/oCuYge
- Davies RJ, Miller R, Coleman N. Colorectal cancer screening: prospects for molecular stool analysis. Nature Rev Cancer. 2005; 5: 199-209. Ref.: https://goo.gl/Pg1myj
- Smith RA, Cokkinides V, Brawley OW. Cancer screening in the United States, 2009. A review of current American Cancer Society Guidelines and issues in cancer screening. CA Cancer J Clin. 2009; 59: 27-41. Ref.: https://goo.gl/KcMdMa
- Centers for Disease Control and Prevention. Increased use of colorectal cancer test: United States, 2002 and 2004,MMWR Mortal Wkly. 2006; 55: 208-311. Ref.: https://goo.gl/dtF1qt
- Ahmed FE. Colon cancer: Prevalence, screening, gene expression and mutation, and risk factors and assessment. J Environ Sci Health C Environ Carcinog Ecotoxicol Rev. 2003; 21: 65-131. Ref.: https://goo.gl/s1THQB
- Morikawa T, Kato J, Yamaji Y, Wada R, Mitsushima T, et al. A Comparison of the immunochemical fecal occult blood test and total colonoscopy in the asymptomatic population. Gastroenterology. 2005; 129: 422-428. Ref.: https://goo.gl/qFWRqy
- Kohler BA, Ward E, McCarthy BJ, Edwards BK, Jemal A, et al. Annual report to the nation on the status of cancer, 1975-2007, featuring colorectal cancer trends and impact of interventions (risk factors, screening, and treatment) to reduce future rates. Cancer. 2010; 116: 544-573. Ref.: https://goo.gl/HEU4v3
- Ahlquist DA. Fecal occult blood testing for colorectal cancer. Can we afford to do this? Gastroenterol Clin N Amer. 1997; 26: 41-55, Ref.: https://goo.gl/CiDZiY
- Davidson LA, Lupton JR, Miskovsky E, Miskovsky, Alan P. Fields, et al. Quantification of human intestinal gene expression profiling using exfoliated colonocytes: a pilot study. Biomarkers. 2003; 8: 51-61. Ref.: https://goo.gl/kam5TN
- Ahmed FE, Jeffries CD, Vos PW, Flake G, Nuovo GJ, et al. Diagnostic microRNA markers for screening sporadic human colon cancer and ulcerative colitis in stool and tissue. Cancer Genom Proteom. 2009; 6: 281-296. Ref.: https://goo.gl/7cKJBE
- Ahmed FE, Vos P, iJames S, Lysle DT, Allison RR, et al. Transcriptomic molecular markers for screening human colon cancer in stool & tissue. Cancer Genom Proteom. 2007; 4: 1-20, 2007. Ref.: https://goo.gl/7yJ3eB
- Ahmed FE, Ahmed NC, Vos PW, Bonnerup C, Atkins JN,et al. Diagnostic microRNA markers to screen for sporadic human colon cancer in stool: I. Proof of principle. Cancer Genom Proteom. 2013; 10:93-113. Ref.: https://goo.gl/FPuKfyc
- Ahmed FE, Ahmed NC, Vos PW, Bonnerup C, Atkins JN, et al. Diagnostic microRNA markers to screen for sporadic human colon cancer in blood. Cancer Genom Proteom. 2012; 9: 179-192. Ref.: https://goo.gl/EDntph
- Ahlquist DA. Fecal occult blood testing for colorectal cancer. Can we afford to do this? Gastroenterol Clin North Am. 1997; 26: 41-55. Ref.: https://goo.gl/htMCvJ
- Cheng L, Eng G, Nieman L, Kapadia AS, Du XL.Trends in colorectal cancer incidence by anatomic site and disease stage in the United States from 1976 to 2005. Am J Clin Oncol. 2011; 34: 573-580. Ref.: https://goo.gl/VkxgTB
- Huxley RR, Ansary-Moghaddam A, Clifton P, Czernichow S, Parr CL, et al.The impact of dietary and lifestyle risk factors on risk of colorectal cancer: a quantitative overview of the epidemiological evidence. J Natl Cancer Inst. 2009; 125: 171-180. Ref.: https://goo.gl/i4eq6N
- Morikawa T, Kato J, Yamaji, Wada R, Mitsushima T, et al. Comparison of the immunochemical fecal occult blood test and total colonoscopy in the asymptomatic population. Gastroenterology. 2005; 129: 422-428. Ref.: https://goo.gl/iHrxpa
- Newcomb PA, Storer BE, Morimoto LA, Templeton A, Potter JD. Long-term efficacy of sigmoidoscopy in the reduction of colorectal cancer incidence. J Natl Cancer Inst. 2003; 95: 622-625. Ref.: https://goo.gl/iAzxbz
- Yamai Y, Mitsushima T, Ikuma H, Watabe H, Okamoto M, et al.Right-sided shift of colorectal adenomas with aging. Gastrointest Endoscopy. 2006;63: 453-458. Ref.: https://goo.gl/ZPW9Cj
- Gatto NM, Frucht H, Sundarararjan V, Jacobson JS, Grann VR, et al. Risk of perforation after colonoscopy or sigmoidoscopy: a population based study. J Natl Cancer Inst. 2003; 95: 230-236. Ref.: https://goo.gl/QzDjNM
- Birkenkamp-Demtroder K, Olesen SH, Sørensen FB, Laurberg S, Laiho P, et al. Differential gene expression in colon cancer of the ceacum versus the sigmoid and rectosigmoid. Gut. 2005; 54: 374-384. Ref.: https://goo.gl/Cv6fVD
- Gervaz P, Bouzourene H, Gerottini JP. Dukes B colorectal cancer: distinct genetic categories and clinical outcome based on proximal or distal tumor locations. Dis Colon Rectum. 2001; 44: 364-372.
- Bressler B, Paszat LF, Vinden C, Li C, He J, et al. Colonoscopic miss rates for right-sided colon cancer: population-based study.Gastroenterology. 2004; 127: 452-456. Ref.: https://goo.gl/gqM49H
- Mulhall BP, Veerappan GR, Jackson J. Meta-analysis: Computed tomographic colonography. Ann Intern Med. 2005; 142: 635-650. Ref.: https://goo.gl/Fhefc9
- Kealey SM, Dodd JD, MacEneaney PM, Gibney RG, Malone DE. Minimal preparation computed tomography instead of barium enema/colonoscopy for suspected colon cancer in frail elderly patients: an outcome analysis study. Clinical Radiol. 2004; 59: 44-52. Ref.: https://goo.gl/pYJV17
- Mȕller H M, Oberwalder M, Fiegl H, Morandell M, Goebel G, et al. Methylation changes in fecal DNA: a marker for colorectal cancer screening. Lancet. 2004; 363: 1283-1285. Ref.: https://goo.gl/uJuByP
- Lenhard K, Bommer GT, Asutay S, Schauer R, Brabletz T, et al. Analysis of promoter methylation in stool: a novel method for the detection of colorectal cancer. Clin Gastroenterol Hepatol. 2005; 3: 142-149. Ref.: https://goo.gl/FJi9r5
- Itzkowitz SH, Jandorf L, Brand R, Rabeneck L, Schroy PC 3rd, et al. Improved fecal DNA test for colorectal cancer screening. Clin Gastroenterol Hepatol. 2007; 5: 111-117. Ref.: https://goo.gl/yh4Ln5
- Imperiale TF, Ransohoff DF, Itzkowitz SH, Turnbull BA, Ross ME, et al. Fecal DNA versus fecal occult blood for colorectal cancer screening in an average-risk population. New Eng J Med. 2004; 351: 2704-2714. Ref.: https://goo.gl/ivNqvJ
- Ahmed FE. Liquid chromatography-mass spectrometry: A tool for proteome analysis & biomarker discovery and validation. Exp Opin Mol Diag. 2009; 3: 429-444. Ref.: https://goo.gl/icM5wn
- Osborn NK, Ahlquist DA. Stool screening for colorectal cancer: molecular approaches. Gastroenterology. 2005;128: 192-206. Ref.: https://goo.gl/ghMA4Q
- Ahlquist DA, Shuber AP. Stool screening for colorectal cancer: evolution from occult blood to molecular markers. Clin Chim Acta. 2002; 315: 151-157. Ref.: https://goo.gl/AJkUr2
- Traverso G, Shuber A, Levin B, Johnson C, Olsson L, et al. Detection of APC mutations in fecal and DNA from patients with colorectal tumors. New Engl J Med. 2002; 346: 311-320. Ref.: https://goo.gl/o5Svd1
- Ahlquist DA, Skoletsky JE, Boynton KA, Harrington JJ, Mahoney DW, et al. Colorectal cancer screening by detection of altered human DNA in stool: feasibility of a multitarget assay panel. Gastroenterology. 2000; 119: 1219-1227. Ref.: https://goo.gl/Zh284Z
- Ladabaum U and Song K. Projected national impact of colorectal cancer screening on clinical and economic outcomes and health services demand. Gastroenterology. 2005; 129: 1151-1126. Ref.: https://goo.gl/KDLhgo
- Polley AC, Mulholland F, Pin C, Williams EA, Bradburn DM, et al. Proteomic analysis reveals field-wide changes in protein expression in the morphologically normal mucosa of patients with colorectal neoplasia. Cancer Res. 2006; 66: 6553-6562. Ref.: https://goo.gl/vER2Ub
- Xin B, Platzer P, Fink SP, Reese L, Nosrati A, et al. Colon cancer secreted protein-2 (CCSP-2) a novel candidate serological marker of colon neoplasia. Oncogene. 2005; 24: 724-731. Ref.: https://goo.gl/WPSSdC
- Thomas SN, Zhu F, Schnaar RL, Alves CS, Konstantopoulos K. Carcinoembryonic antigen and CD44 variant isoforms cooperate to mediate colon carcinoma cell adhesion to E- and L-selectin in shear flow. J Biol Chem. 2008; 283, 15647-15655. Ref.: https://goo.gl/xRW845
- Koprowski H, Herlyn M, Steplewski Z, Sears HF. Specific antigen in serum of patients with colon carcinoma. Science. 1981; 212: 53-55. Ref.: https://goo.gl/X55j65
- Smith RA, von Eschenbach AC, Wender R, et al. American Cancer Society guidelines for the early detection of cancer: update of the early detection guidelines for prostate, colorectal and endometrial cancers. CA Cancer J Clin. 2001; 51: 38-75.
- Ng EKO, Chong WWS, Jin H, Lam EK, Shin VY, et al. Differential expression of microRNA in plasma of patients with colorectal cancer: A potential marker for colorectal cancer screening. Gut. 2009; 58: 1375-1381.Ref.: https://goo.gl/vdrg7u
- Link A, Balaguer F, Shen Y, Nagasaka T, Lozano JJ, et al. Fecal miRNAs as novel biomarkers for colon cancer screening. Cancer Epidemiol Biomarkers Prev. 2010; 19: 1766-1774. Ref.: https://goo.gl/T8zM6n
- Koga Y, Yasunaga M, Takahashi A, Kuroda J, Moriya Y, et al. MicroRNA expression profiling of exfoliated colonocytes isolated from feces for colorectal cancer screening. Cancer Prev Res. 2010; 3: 1435-1442. Ref.: https://goo.gl/omh9cj
- Kalimutho M, Del Vecchio BG, Di Cecilia S, Sileri P, Cretella M, et al. Differential expression of miR-144* as a novel fecal-based diagnostic marker for colorectal cancer. J Gastroenterol. 2011; 46: 1391-1402. Ref.: https://goo.gl/8ofmSf
- Kalimutho M, Di Cecilia S, Del Vecchio BG, Roviello F, Sileri P, et al. Epigenetically silenced miR-34b/c as a novel faecal-based screening marker for colorectal cancer. Br J Cancer. 2011; 24: 1770-17780. Ref.: https://goo.gl/S6BHd2
- Kunte DP, Delacruz M, Wali RK, Menon A, Du H, et al. Dysregulation of microRNAs in colonic field carcinogenesis: implications for screening. PLoS One. 2012; 7. Ref.: https://goo.gl/9Uv87v
- Wu CW, Ng SS, Dong YJ, Ng SC, Leung WW, et al. Detection of miR-92a and miR-21 in stool samples as potential screening biomarkers for colorectal cancer and polyps. Gut. 2012; 61: 739-745. Ref.: https://goo.gl/KM4KTC
- Cummins JM, He Y, Leary RJ, Pagliarini R, Diaz LA Jr, et al. The colorectal microRNome. Proc Natl Acad Sci USA. 2006; 103: 3687-3692. Ref.: https://goo.gl/1ffTMh
- Schepler T, Reinert JT, Oslenfeld MS, Christensen LL, Silahtaroglu AN, et al. Diagnostic and prognostic microRNAs in Stage II colon cancer. Cancer Res. 2008; 68: 6416-6424. Ref.: https://goo.gl/7jxmdN
- Barbarotto E, Schmittgen TD, Calin GA. MicroRNAs and cancer: Profile, profile, profile. Int J Cancer. 2008; 122: 969-977. Ref.: https://goo.gl/AAH3YA
- Schetter AJ, Leung SY, Sohn JJ, Harris HH, Calin GA, et al. MicroRNA expression profile associated with progression and therapeutic outcome in colon adenocarcinoma. J Am Med Assoc. 2008; 299: 425-436. Ref.: https://goo.gl/rG9qaK
- Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006; 6: 857-866. Ref.: https://goo.gl/a4VaZA
- Lu J, Getz G, Miska EA, Eric A, Alvarez-Saavedra, Ezequiel, et al. MicroRNA expression profiles classify human cancers. Nature. 2005; 435: 834-838. Ref.: https://goo.gl/2tBaAP
- Yantis RK, Goodarzi M, Zhou XK, Rennert H, Pirog EC, et al Clinical, pathological, and molecular features of early-onset colorectal carcinoma. Am J Surg Pathol. 2009; 33: 572-582. Ref.: https://goo.gl/Pkcs1N
- Luo X, Burwinke B, Tao S, Brenner J. MicroRNA signatures: Novel biomarkers for colorectal cancers. Cancer Epidemiol Biomarkers Prev. 2011; 20: 1272-1286. Ref.: https://goo.gl/rz2aXY
- Ahmed FE Testing for genetically modified organisms (GMOs) in food products. Lab Plus Intern. 2002; 16: 8-16.
- Ahmed FE, Vos P Molecular markers for human colon cancer in stool and blood identified by RT-PCR. Anticancer Res. 2004; 24: 4127-4134.
- Wang K, Zhang S. Weber J, Baxter D, Galas DJ. Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res. 2010; 38: 7248-7259. Ref.: https://goo.gl/7Z4k68
- Arroyo JD, Chevillet JR, Kroh EM, Ruf IK, Pritchard CC, et al. Argonaute 2 complexes carry a population of circulating microRNAs independent of vesicles in human plasma. Proc Natl Acad Sci USA. 2011; 108: 5003-5008. Ref.: https://goo.gl/6ithpn
- Vickers KC, Palmisano BT, Shoucri BM, Shamburek RD, Remaley AT. MicroRNAs are transported in plasma and delivered to recipient cells by high-density lipoproteins. Nat Cell Biol. 2011; 13: 423-433. Ref.: https://goo.gl/LVkXDu
- Hunter MP. Detection of microRNA expression in human peripheral blood microvessicles. PLoS One. 2008; 3: e3694. Ref.: https://goo.gl/VCsF6q
- Shaffer J, Schlumpberger M, Lader E. miRNA profiling from blood-Challenges and recommendations. 2012; 1-10. Ref.: https://goo.gl/UzZJcA
- Ahmed FE, James SI, Lysle DT, Johnke RM, Flake G, et al. Improved methods for extracting RNA from exfoliated human colonocytes in stool and RT-PCR analysis. Dig Dis Sci. 2004; 49: 1889-189. Ref.: https://goo.gl/ZL3RF1
- Mestdagh P, Van Vlierberghe P, Weer De, Muth D, Westermann F, et al. A novel and universal method for microRNA RT-qPCR data normalization. Genome Biology. 2009; 10: R64. Ref.: https://goo.gl/NcZsbM
- Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet. 2005; 37: 766-770. Ref.: https://goo.gl/SdnC8n
- Balcells I, Cirera S, Busk PK. Specific and sensitive quantitative RT-PCR of miRNAs with DNA primers. BMC Biotechnol. 2011; doi: 10.1186/1472-6750-11-70. Ref.: https://goo.gl/acP9sf
- Resnick KE, Alder H, Hagan JP, Richardson DL, Croce CM, et al. The detection of differentially expressed microRNAs from the serum of ovarian cancer patients using a novel real-time PCR platform. Gynecol Oncol. 2009; 112: 55-59. Ref.: https://goo.gl/r3FHeQ
- Redshaw N, Wilkes T, Whale A, Cowen S, Huggett J, et al. A comparison of miRA isolation and RT-qPCR technologies and their effects on quantification accuracy and repeatability. BioTechniques. 2013; 54: 155-164. Ref.: https://goo.gl/odZL4n
- Lee EJ, Gusev Y, Jiang J, Nuovo GJ, Lerner MR, et al. Expression profiling identifies distinct microRNA signature in pancreatic cancer. Int J Cancer.2007; 120: 1046-1054. Ref.: https://goo.gl/GkjLKn
- Yanaihara N, Caplen N, Bowman E, Seike M, Kumamoto K, et al. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell. 2006; 9: 189-198. Ref.: https://goo.gl/iaXvCV
- Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005; 65: 7065-7070. Ref.: https://goo.gl/gRnTYF
- Miska EA, Alvarez-Saavedra E, Townsend M, Yoshii A, Rakic P, et al. Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol. 2004;5: R68. Ref.: https://goo.gl/aGnWQi
- Kim J, Krichevsky A, Grad Y, Gabriel D, Kenneth S, et al. Identification of many microRNAs that copurify with polyribosomes in mammalian neurons. Proc Natl Acad Sci USA. 2004; 101: 360-365. Ref.: https://goo.gl/g4rd6K
- Volinia S, Calin GA, Liu CG, Cimmino A, Petrocca F, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci USA. 2006; 103: 2257-2261. Ref.: https://goo.gl/tdtGu8
- Aandrés E, Cubedo E, Agirre X, Malumbres R, Navarro A, et al. Identification by real-time PCR of 13 mature microRNAs differentially expressed in colorectal cancer and non-tumor tissues. Mol Cancer. 2006;5: 29. Ref.: https://goo.gl/QsXhHD
- Jiang J, Lee EJ, Gusev Y, Schmittgen TD. Real-time expression profiling of microRNA precursors in human cancer cell lines. Nucleic Acids Res. 2005; 33: 5394-5403. Ref.: https://goo.gl/hYNyXD
- Shi B, Stepp-Lorenzino L, Prisco M, Linsley P, Baserga R, et al. MicroRNA 145 targets the insulin receptor substrate-1 and inhibits the growth of colon cancer cells. J Biol Chem. 2007; 282: 32582-32590. Ref.: https://goo.gl/fx31Av
- Calin GA, Ferracin M, Cimmino A, Shimizu M, Visone R, et al. A microRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Eng J Med. 2005; 353: 1793-1801. Ref.: https://goo.gl/9xaxAH
- Eis PS, Tam W, Sun L, Chadburn A, Li Z, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci USA. 2003; 102: 3627-3632. Ref.: https://goo.gl/3tiZ6j
- Calin GA, Dumitru CD, Shimizu M, Bichi R, Zupo S, et al. Frequent deletions and downregulation of microRNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acas Sci USA. 2002; 99: 15524-15529. Ref.: https://goo.gl/k8rtMh
- Nybo K, Lo PCH. Optimal miRNA RT-qPCR. BioTechniques. 2013;54: 113.
- Ahmed FE, Vos PW, Clark J, Wiley JE, Weidner DA, et al. Differences in mRNA and microRNA expression profiles in human colon adenocarcinoma HT-29 cells treated with either intensity-modulated radiation therapy (IMRT), or conventional radiation therapy (RT). Cancer Genom Proteom. 2009; 6: 109-127. Ref.: https://goo.gl/PQodJ6
- Wu F, Zikusoka M, Trindade A, Dassopoulos T, Chakravarti S, et al. MicroRNAs are differentially expressed in ulcerative colitis and alter expression of macrophage inflammatory particle 2-α. Gastroenterology. 2008; 135: 1626-1635. Ref.: https://goo.gl/edUJbv
- Lu M, Zhang Q, Deng M, Miao, Cui Q, et al. An analysis of human microRNA and disease associations. PLoS One. 2008; 3: e3420. Ref.: https://goo.gl/YQYE4V
- Ahmed FE Expression microarray proteomics and the search for cancer biomarkers. Curr Genomics. 2006; 7: 399-426. Ref.: https://goo.gl/fvhGMA
- Ahmed FE. Quantitative real-time RT-PCR: Application to carcinogenesis. Cancer Genom Proteom. 2005; 2: 317-332. Ref.: https://goo.gl/6WZE5f
- Lewis BP, Shih IH, Jones-Rhodes MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003; 115: 787-789. Ref.: https://goo.gl/EQCSzC
- Gusev Y. Computational methods for analysis of cellular functions and pathways collectively targeted by differentially expressed microRNA. Methods. 2008; 44: 61-72. Ref.: https://goo.gl/BgFKoA
- Gusev Y, Schmittgen TD, Lerner M, Postier R, Brackett D. Computational analysis of biological functions and pathways collectively targeted by coexpressed microRNAs in cancer. BMC Bioinformatics. 2007; 8(Suppl 7): S16. Ref.: https://goo.gl/4wsGK5
- Ahmed FE. The role of microRNA in carcinogenesis and biomarker selection: a methodological perspective. Exp Rev Mol Diag. 2007; 7: 569-603. Ref.: https://goo.gl/krV5s9
- Sobin LH, Wittekind CH. eds UICC TNM Classification of Malignant Tumors, 6th Edition. New York, John Wiley. 2002; 170-173.
- Greene FL, Page DL, Fleming ID. Eds AJCC Cancer Staging Manual. 6th Edition. Springer-Verlag, New York. 2002.
- DeBakey ME, Yang L, Belaguli N. MicroRNA and colorectal cancer. World J Surg. (2009); 33: 638-646. Ref.: https://goo.gl/pHHkda
- Zhou X, Ruan J, Wang G, Zhang W. Characterization and identification of microRNA core promoters in trout model species. PLoS Comput Biol. 2005; 3: e37. Ref.: https://goo.gl/QeaCdV
- Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNA using deep-sequencing data. Nucleic Acids Res. 2014; 42: D68-D73. Ref.: https://goo.gl/725cpz
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism and function. Cell. 2004;116: 281-297. Ref.: https://goo.gl/dzyaTz
- Reinhart BJ, Slack FJ, Basson M et al RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000; 403: 901-906. Ref.: https://goo.gl/t4bGUJ
- Xu P, Guo M, Hay BA. MicroRNAs and the regulation of cell death. Trend Genet. 2004; 20: 617-624. Ref.: https://goo.gl/rek2Ao
- Chang-Zheng C. MicroRNAs as oncogenes and tumor supressors. N Eng J Med. 2005; 353: 1768- 1771. Ref.: https://goo.gl/1jXDzK
- Calin GA, Sevignai C, Dumitru CD, Hyslop T, Noch E, et al. Human microRNAs are frequently located at fragile sites and genomic regions involved in cancer. Proc Natl Acad Sci USA. 2004; 101: 2999-3004. Ref.: https://goo.gl/8S2Ut9
- Ahmed FE. Molecular markers that predict response to colon cancer therapy. Exp Rev Mol Diag. 2005; 5: 353-375. Ref.: https://www.ncbi.nlm.nih.gov/pubmed/15934813
- Lanza G, Ferracin M, Gafà R, Veronese A, Spizzo R, et al. mRNA/microRNA gene expression profile in microsatellite unstable colorectal cancer. Molecular Cancer. 2007; 6: 54. Ref.: https://goo.gl/EKzZ3r
- Kiriakidou M, Nelson PT, Kouranov A, Fitziev P, Bouyioukos C, et al. A combined computational-experimental approach predicts human microRNA targets. Genes Dev. 2004; 18: 1165-1178. Ref.: https://goo.gl/fipMMv
- John BB, Enright AJ, Aravin A, Tuschl T, Sander C, et al. Human microRNA target. PloS Biol. 2004; 2: e363. Ref.: https://goo.gl/2mFVB9
- Krek A, Grun D, Poy MN, Wolf R, Rosenberg L, et al.Combinational microRNA target predictions. Nature Genet. 2005; 37: 495-500. Ref.: https://goo.gl/4zo78L
- Stark A, Brennecke J, Bushati N, Russell RB, Cohen SM. Animal microRNAs confers robustness to gene expression and have a significant impact on 3’UTR evaluation. Cell. 2005; 123: 1133-1146. Ref.: https://goo.gl/p3Gvna
- Oberg AL, French AJ, French AJ, Subramanian S, Morlan BW et al. MiRNA expression in colon polyps provide evidence for a multihit model of colon cancer. PLoS ONE. 2011; 6: e20465. Ref.: https://goo.gl/h6uvVY
- Valadi H, Elkstrom K, Bossios A,Sjöstrand M, Lee JJ, et al. Exosome mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol. 2007; 9: 654-659. Ref.: https://goo.gl/hPT9bm
- Ahmed FE. Laser microdissection: application to carcinogenesis. Cancer Genom. Proteom. 2006; 3: 217-226.
- Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inbibitors. Proc Natl Acas Sci USA. 1977; 74: 5463-5467. Ref.: https://goo.gl/dTJRbo
- Morozova O, Marra MA. Application of next-generation sequencing technologies in functional genomics. Genomics. 2008; 92: 255-264. Ref.: https://goo.gl/n7gTfz
- Ewing B, Green P. Base-calling of automated sequencer traces using phred. II. Error probabilities. Genome Res. 1988; 8: 186-194. Ref.: https://goo.gl/aefkk9
- Margulies M, Engholm M, Altman WE, Attiya S, Bader JS, et al. Genome sequencing in microfabricated high-density picoliter reactors. Nature. 2005; 437: 376-380. Ref.: https://goo.gl/8txWVi
- Bentley DR. Whole-genome re-sequencing. Curr Opin Genet Dev. 2006; 16: 545-552. Ref.: https://goo.gl/rnJPes
- Shendure J, Porreca GJ, Reppas NB, Lin X, McCutcheon JP, et al. Accurate multiplex polony sequencing at an evolved bacterial genome. Science. 2005; 309: 1728-1732. Ref.: https://goo.gl/QMz8kk
- Jensen SG, Lamy P, Rasmussen MH, Ostenfeld MS, Dyrskjøt L, et al. Evaluation of two commercial global miRNA expression profiling platforms for detection of less abundant miRNAs. BMC Genomics. 2011; 12: 435. Ref.: https://goo.gl/pSZgPA
- Chen C, Ridzon DA, Broomer AJ, Zhou Z, Lee DH, et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005; 33: e179. Ref.: https://goo.gl/U6PHMc
- Tellman G. The E-method: a highly accurate technique for gene-expression analysis. Nature Methods. 2006; 3: 1-2.
- Light Cycler Software®, Version 3.5, Roche Molecular Biochemicals, Mannheim, Germany, 2001; 64-79.
- Luu-The V, Paquet N, Calvo E, Cumps J. Improved real-time RT-PCR method for high-throughput measurements using second derivative calculation and double correction. Biotechniques. 2005; 38: 287-293. Ref.: https://goo.gl/uTwGx8
- Thellin O, Zorzi W, Lakaye B, De Borman B, Coumans B, et al. Housekeeping genes as internal standards: use and limits. J Biotechnol. 1999; 75: 291-295. Ref.: https://goo.gl/CHj4EB
- Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, et al. Accurate normalization of real-time quantitative RT-PCR by geometric average of multiple internal control genes. Genome Biol. 2002; 3. Ref.: https://goo.gl/ywZczE
- DeMuth JP, Jackson CM, Weaver DA, Erin L Crawford, Dennis S, et al. The gene expression index cmyc x E2F-1/p21 is highly predictive of malignant phenotype in human bronchial epithelial cells. Am J Respir Cell Mol Biol. 1998; 19: 18-29. Ref.: https://goo.gl/YH5yrc
- Nagan CY, Yamamoto H, Seshimo I, Ezumi K, Terayama M, et al. A multivariate analysis of adhesion molecules expression in assessment of colorectal cancer. J Surg Oncol. 2007; 95: 652-662. Ref.: https://goo.gl/jKs2S9
- Pepe MS, Feng Z, Janes H, Bossuyt PM, Potter JD. Pivotal evaluation of the occurance of a biomarker used for classification or prediction: standards for study design of Cancer. J.Natl Cancer Inst. 2008; 100: 1432-1438. Ref.: https://goo.gl/Z7zdpA
- Ein-Dor L, Zuk O, Domany E. Thousands of samples are needed to generate a robust gene list for predicting outcome in cancer. Proc Natl Acad Sci USA. 2006; 103: 5923-5928. Ref.: https://goo.gl/koxRNC
- Schwarzenbach H, da Silva A A, Calin G, Pantel K. DNA normalization strategies for microRNA quantification. Clinical Chem. 2015; 61: 1333-1342. Ref.: https://goo.gl/9LB8DZ
- Bustin SA, ed. A-Z of Quantitative PCR. International University Line, La Jolla, CA, 2004.
- Yau TO, Wu CW, Dong Y, Tang CM, Ng SS, et al. MicroRNA-221 and microRNA-18a identification in stool as biomarkers for the non-invasive diagnosis of colorectal carcinoma. Br J Cancer. 2014; 111: 1765-1771. Ref.: https://goo.gl/AUAvH2
- Tichopad A, Dilger M, Schwarz G, Pfaffl MW. Standardised determination of real-time PCR efficiency from a single reaction setup. Nucleic Acids Res. 2003; 31. Ref.: https://goo.gl/eNTPgw
- Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, et al. The MIQUE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin Chem. 2009; 55: 611-622. Ref.: https://goo.gl/fhjcRN
- Cornell RG, Ed. Statistical models for cancer studies. In Models to Analyze Strategies in the General Population, 346-347, Marcel Dekker, NY, 1984.
- Sureh KP, Chandrashekare S. Sample size estimation and power analysis for clinical research studies. J Hum Reprod Sci. 2012; 5: 7-13. Ref.: https://goo.gl/UDoWY6
- Moore DS, McCabe GP, Craig B. Introducrion to the Practice of Statistics, 6th edition. W.H. Freeman & Company, St. Louis, MO, 2009.
- Tang Y, Ghosal S, Roy A. Nonparametric Bayesian estimation of positive false discovery rates. Biometrics. 2007; 63: 1126-1134. Ref.: https://goo.gl/fhT11d
- Nagan CY, Yamamoto H, Seshimo I, Ezumi K, Terayama M, et al. A multivariate analysis of adhesion molecules expression in assessment of colorectal cancer. J Surg Oncol. 2007; 95: 652-662. Ref.: https://goo.gl/5Wr9zQ
- Yildiz OY, Aslan A, Alpagdin E. Multivariate statistical tests for comparing classification algorithms. In Learning and Intelligence Optimization. Springer. 2011.
- Reiner A, Yekutieli D, Benjamini Y. Identyfying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics. 2003; 19: 368-375. Ref.: https://goo.gl/zdjwUe
- Pawitan Y, Michiels S, Kosciely S, Gusnato A, Polner A. False discovery rate, sensitivity and sample size for microarray studies. Bioinformatics. 2005; 21: 3017-3024. Ref.: https://goo.gl/MDCtsB
- Choi H, Nesvizhskii AI. False discovery rates and related statistical concepts in mass spectrometry-based proteomics. J Proteome Res. 2008; 7: 47-50. Ref.: https://goo.gl/jEKHGT
- Earl-Slatter A. Cross Validation, In the Handbookmof Clinical Trials and Other Research. Radcliff Medical Press Ltd. 2002.
- Efron B, Tibshirani RJ. An introduction to the Bootstrap, Chapman and Hall. 1993.
- Hanley JA, McNeil BJ. The meaning and use of the area under a receiver operating characteristic (ROC) curve. Radiology. 1982; 143: 29-36. Ref.: https://goo.gl/rWnWhk
- Ringer M. What is principal component analysis? Nature Biotechnol. 2008; 26: 303-304. Ref.: https://goo.gl/F3nW3g
- Wegman E. Hyperdimensional data analysis using parallel coordinate. J Am Stat Assoc. 1990; 85: 644-675. Ref.: https://goo.gl/rZjzfv
- Gabriel KR, Odoroff CL. Biplots in biomedical research. Stat Med. 1990; 9: 469-485. Ref.: https://goo.gl/BCWcBJ
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protocol. 2009; 4: 44-57. Ref.: https://goo.gl/Ef9K2k
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserveds tissue-specific CpG island shores. Nat Genet. 2009; 41: 178-186. Ref.: https://goo.gl/5tpHH5
- Herman JG, Baylin SB. Gene silencing in association with promoter hypermethylation. N Eng J Med. 2003; 349: 2042-2054. Ref.: https://goo.gl/CwmWdN
- Hansen KD, Timp W, Corrada H, Sabunciyan S, Langmead B, et al. Increased methylation variation in epigenetic domains across cancer types. Nature Genet. 2011; 43: 768-775. Ref.: https://goo.gl/d4heLS
- Sarver AL, French AJ, Borralho PM, Thayanithy V, Oberg AL, et al. Human colon cancer profiles show differential microRNA expression depending on mismatch repair status and are characteristic of undifferentiated proliferative states. BMC Cancer. 2009; 9: 401. Ref.: https://goo.gl/r48pYJ
- Earle JS, Luthra R, Romans A, Abraham R, Ensor J, et al. Association of microRNA expression with microsatellite instability status in colorectal adenocarcinoma. J Mol Diag. 2010; 12: 433-440. Ref.: https://goo.gl/Mzn7aC
- Balaguer F, Moreira L, Lozano JJ, Link A, Ramirez G, et al. Colorectal cancers with microsatellite instability display unique miRNA profiles. Clin Cancer Res. 2011; 17: 6239-6249. Ref.: https://goo.gl/Xj5m2D
- Tuddenham L, Wheeler G, Ntounia-Fousara S, Waters J, Hajihosseini MK, et al. The cartlidge specific microRNA-140 targets histone deacetylase 4 in mouse cells. FEBS Lett. 2006; 580: 4214-4217. Ref.: https://goo.gl/uCPD9h
- Costa Y, Speed RM, Gautier P, Semple CA, Maratou K, et al. Mouse MAELSTROM: the link between miotic silencing of unsynapsed chromatin and microRNA pathways? Hum Mol Genet. 2006; 15: 2324-2334. Ref.: https://goo.gl/g5ApSb
- Rajewsky N. microRNA target predictions in animals. Nat Genet. 2006; 38: S8-S13. Ref.: https://goo.gl/1Y3BRr
- Saito Y, Liang G, Egger G, Friedman JM, Chuang JC, et al. Specific activation of microRNA-127 with downregulation of the protooncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006; 9: 435-443. Ref.: https://goo.gl/4CvfGe
- Lujambio A, Calin GA, Villanueva A, Ropero S, Sánchez-Céspedes M, et al. A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci USA. 2008; 105: 13556-13561. Ref.: https://goo.gl/czoFWV
- Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, et al. Augmentation of tumor angiogenesis by a myc-activated microRNA cluster. Nature Genet. 2006; 38: 1060-1065. Ref.: https://goo.gl/P2kXXb
- Fazi F, Rosa A, Fatica A, Gelmetti V, De Marchis ML, et al. A minicircuitry comprised of microRNA-223 and transcription factors NFI-A and C/EBPalpha regulates human granulopoiesis. Cell. 2005; 123: 819-831. Ref.: https://goo.gl/c2ogai
- Koss LG, Melamed MR, Eds. Koss’ Diagnostic Cytology and Histopathologic Bases, 5th edition, Lippincott, Williams & Wilkins, 2005.
- Winter MJ, Nagtegaal ID, van Krieken JH, Litvinov SV. The epithelial cell adhesion molecule (Ep-CAM) as a morphoregulatory molecule is a tool in surgical pathology. Am J Pathol. 2003; 163: 2139-2148. Ref.: https://goo.gl/9qiDgf
- Petrelli NJ, Letourneau R, Weber T, Nava ME, Rodriguez-Bigas M. Accuracy of biopsy and cytology for the preoperative diagnosis of colorectal adenocarcinoma. J Surg Oncol. 1999; 71: 46-49. Ref.: https://goo.gl/KsPfWB
- Matsushita HM, Matsumura Y, Moriya Y, Akasu T, Fujita S, et al. A new method for isolating colonocytes from naturally evacuated feces and its clinical application to colorectal cancer diagnosis. Gastroenterology. 2005; 129: 1918 - 1927. Ref.: https://goo.gl/mZAy84
- Simpson RJ, Lim JE, Moritz RL, Mathivanan S. Exosomes: proteomic insights and diagnostic potential. Expert Rev Proteomics. 2009; 6: 267-283. Ref.: https://goo.gl/RR1Xtf
- Baker M. Digital PCR hits its stride. Nature Methods. 2012; 9: 541-544. Ref.: https://goo.gl/FQfNH5
- McShane LM, Altman DG, Sauerbrei W, Sheila E. Taube, Massimo Gion, et al. Reporting recommendations for tumor marker prognostic studies (REMARK). J Natl Cancer Inst. 2005; 97: 1180-1184. Ref.: https://goo.gl/nGLTAy