Research Article
The master regulator gene PRDM2 controls C2C12 myoblasts proliferation and Differentiation switch and PRDM4 and PRDM10 expression

Di Zazzo Erika#, Bartollino Silvia*# and Moncharmont Bruno
Department of Medicine and Health Sciences “V. Tiberio”, University of Molise, Italy
#These authors contributed equally to this work
*Address for Correspondence: Silvia Bartollino, Department of Medicine and Health Sciences, “V. Tiberio”, University of Molise Via F. De Sanctis, 86100 Campobasso, Italy, Tel.: +39 0874404886, E-mail: [email protected]
Dates: Submitted: 11 August 2017; Approved: 20 September 2017; Published: 25 September 2017
How to cite this article: Di Zazzo E, Bartollino S, Moncharmont B. The master regulator gene PRDM2 controls C2C12 myoblasts proliferation and Differentiation switch and PRDM4 and PRDM10 expression. Insights Biol Med. 2017; 1: 075-091. DOI: 10.29328/journal.ibm.1001007
Copyright License: © 2017 Di Zazzo E, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: PRDM; RIZ; Cell cycle; Proliferation; Differentiation
Abstract
The Positive Regulatory Domain (PRDM) protein family gene is involved in a spectrum variety of biological processes, including proliferation, differentiation and apoptosis: its member seem to be transcriptional regulators highly cell type and tissue peculiar, towards histones modifications or recruitment of specific interaction patters to modify the expression of target genes. In this study we analyzed the expression profile of different member of PRDM gene family focusing our attention on the role of PRDM2, PRDM4 and PRDM10 genes in mouse C2C12 cell line, during the differentiation of myoblasts into myotubes and speculate about the role of the protein Retinoblastoma protein-interacting zinc finger protein 1-RIZ1, coded by PRDM2 gene, as a regulator of the proliferation/differentiation switch.
Results showed a reduction of PRDM2, PRDM4 and PRDM10 expression level during the commitment of the differentiation of myoblasts into myotubes. The RIZ1 silencing stimulated myoblasts differentiation, similar to the effect of serum deprivation on these cells, associated with an increase of Myogenin expression level, which is considered to be involved in the differentiation of myoblasts into multinucleated myotubes. As demonstrated by chromatin immunoprecipitation experiments, RIZ1 is associated with Myogenin promoter in proliferation condition and after 24h from differentiation induction, negatively controlling therefore Myogenin expression. Moreover RIZ1 silencing induced a reduction in PRDM4 and PRDM10 expression levels leaving us to speculate that the PRDM genes have a redundant role and they are hierarchically organized.
Introduction
The myogenesis of mammalian skeletal muscle cells is regulated by myogenic regulatory factors (MRFs), related in a hierarchical relationship [1], which are considered to be members of a superfamily of the highly conserved variant of the basic helix-loop-helix (bHLH) domain [2], that confers their peculiar myogenic potential [3]. The phylogenetic analysis of the sequences of these genes indicates that MRF genes have evolved from a single ancestral MRF gene progenitor, by gene duplication events followed by divergent mutations [4]. The four main basic helix-loop-helix myogenic regulatory factors, which exhibit a pivotal role in skeletal muscle development and which are responsible for coordinating muscle-specific gene expression in the developing embryo [5], are MyoD (Myf-3) [3], which coordinates an open chromatin structure at muscle-specific genes [1]; Myf-5 [6], which is required for the specification and proliferation of myoblasts [7-10], enhances myogenesis by promoting myoblast proliferation [11-13], because it is also the earliest to be expressed during myogenesis process, acting as a transcription factor in muscle progenitor cells (satellite cells) and myocytes [14]; Myogenin (Myf-1) [15], which is involved in the differentiation of myoblasts into multinucleated myotubes [16] and MRF4 [17], which is implicated in the latest phase of differentiation that includes myofiber maintenance [18]. The Myogenic regulatory factor (MRF) genes are expressed with a peculiar pattern [19,20] and they can auto- and cross-regulate the expression of each other and interact with the myocyte enhancer factor-2 (MEF2) family of transcription factors, to activate the transcription of muscle-specific genes [21]: currently, these MEF2 transcription factors are considered to drive the development of muscle, cardiac, skeletal, vascular, neural and blood cells, because of their pivotal effects on cell differentiation, proliferation, apoptosis, migration, shape and metabolism [22].
The activity of MRF proteins require heterodimerization with a member of the ubiquitously expressed E-protein family of bHLH proteins. This event leads the binding to the regulatory regions of muscle-specific genes on the E-box consensus sequence (CANNTG) [5]. Currently, only a small fraction of 14 million potential sites are available for MRF binding [23,24]. MyoD, the master regulator of the skeletal muscle gene expression program, activates genes which display the consensus E-box sequence VCASCTG (where V is A, C or G while S is C or G) within their promoter/enhancer regions [23,25-27] and it leads a gene expression program by heterodimerizations with E-proteins and giving rise to multinucleated myotubes [28]. The binding of MyoD on the E-box sequence (CANNTG) and the recruitment of factors, which are involved in remodelling the chromatin, are considered the crucial event for transcription [29,30] because MyoD activates pRb and p21 gene expression [31,32] to shoot down the cell cycle machinery [29]. The skeletal muscle differentiation is a strongly coupled event to the cell cycle exit [33] by an upregulation of cyclin-dependent kinase inhibitors (CDKIs), which inhibit cyclin-CDK complexes driving a downregulation of the activity of cyclin D1,E,A, and B-CDK complexes [34] and by an induction of the permanent cell cycle exit. This is a pivotal step because of the overexpression of cyclin/CDKs has been reported to inhibit the activity of MyoD by different mechanisms [29]. In particular, p21cip1/waf1, and p57/kip2, encoded respectively by Cdkn1a and Cdkn1c genes, control differentiation of skeletal muscle and their loss affects fiber formation: they have a key role both for cell cycle exit, both in triggering a muscle-specific transcriptional program [35]. The PRDM (Positive Regulatory Domain) gene family which consists of 17 orthologs in primates and 16 orthologs in rodents, birds and amphibians [36], encodes transcription factors with a PR domain and a variable number of zinc finger motifs [37], with the exception of PRDM11 [36,38]. The PR domain displays a 20-30% amino acid homology sequence to the catalytic SET (Suvar3-9, Enhancer-of-zeste, Trithorax) domain with hystone lysine methyltransferase (HMTs) activity [39]. In contrast to the SET domain proteins, currently only three PRDM proteins have been demonstrated to possess intrinsic HMTase activity [40] and a number of PRDMs have not endowed with catalytic activity towards histones/nucleosomes [41-44]. In particular the HMT’s activity has been found only in the PR domains of PRDM2/RIZ1, Prdm8 and Prdm9 [45-47]. In fact, the PR domain has diverged significantly from the SET domain [48-51] and most PR domains lack the H/RxxNHxC motif required for methyltransferase activity [52,53]. Generally, the PR domain is localized at the N-terminus of the protein, whereas the SET domain is often localized to the C-terminus [39]. A common characteristic of PRDM genes is the expression of molecular variants by alternative splicing or by alternative use of promoters. The PRDM1, PRDM2 and PRDM3, which is also called MECOM (MDS1-EVI1 complex locus) genes are expressed as two alternative forms, by intergenic splicing, which produce the PR plus and PR minus forms of these genes [54-57]. PR plus and PR minus forms are expressed at equimolar concentration and their ratio is maintained in a fine equilibrium [58]: an imbalance in the amounts of the two products, through either disruption or underexpression of the PR plus form or overexpression of the PR minus form commonly occurs in human cancers through genetic and epigenetic mechanisms [59-66]. PRDM2 gene gives two alternative products: RIZ1, the PR plus form, implicated in tumor suppressor function, and RIZ2, the PR minus form. This PR domain (PRDI-BF1 and RIZ homologous) [67] which is endowed with histone H3 K9 methyltransferase activity, is targeted by inactivating mutations in human cancers [68].
PRDM proteins mediate transcriptional activation or repression depending on the nature of their intrinsic HMT activity: PRDM proteins appear to function by modulating gene expression states either directly (via intrinsic HMTase activity), or indirectly (via recruitment of various cofactors), controlling critical aspects of cell integrity, spanning from cell differentiation to cell growth and apoptosis [58]. These genes also play a role in human cancer, where they mainly act as tumor suppressors: for example, PRDM1 is a tumor suppressor of diffuse large B cell lymphoma (DLBCL); PRDM3and PRDM16 show different isoforms with separate functions in leukemia [44,69]. PRDM16 (PRD1-BF1-RIZ1 homologous domain containing 16) controls a bidirectional cell fate switch between skeletal myoblasts and brown fat cells [70], the targeting of 3’UTR of Prdm16 is involved in the choise between myogenic and brown adipose determination of the adult skeletal muscle stem cells (satellite cells) [71], PRDM5 acts with a potential tumor suppressor role for gastrointestinal carcinogenesis [72-74].
The RIZ proteins regulate cell proliferation in a yin-yang manner [57,58,75]: gene silencing of the RIZ1 form, by genetic or epigenetic mechanisms, has been described in a variety of human tumors [14], whereas the RIZ2 form, lacking of PR domain, is always present or overexpressed [76]: this finding suggests a positive selection for RIZ2 in cancer progression. Further evidence indicates that forced expression of RIZ1 in tumor cell cultures induces growth arrest and apoptosis, possessing an anticancer activity in the PR domain [77], and the silencing of RIZ1 expression can stimulate breast cancer cell proliferation [78]. In addition, forced expression of the Zn-finger domain present in both RIZ forms increases the growth rate of breast cancer cells [79]. Based on these findings, RIZ1 could be considered as a crucial tumor suppressor gene candidate and the Zn-finger domain could be responsible for the putative oncogenic activity of the RIZ2 gene product. Such an effect might be more relevant in estrogen target tissues, where RIZ gene products are reported to directly interact with the estrogen receptor in a hormone-dependent manner through a LXXLL motif [80,81], promoting optimal estrogen response; conversely, in osteosarcoma cancer cell line (SAOS2), RIZ1 is expressed at high level in proliferating cell compared to serum-free culture conditions [82]. Murine models studies demonstrated that PRDM1 regulates commitment, as well PRDM14, PRDM9 is involved in meiosis [83], PRDM16 is involved in the switch controlling of myoblast differentiation to brown adipocytes [70,84]; PRDM3 and PRDM16 are also required for the maintenance of brown adipocyte identity [85], but currently the PRDM2 role in the development and in myoblast differentiation is still unclear. The purpose of this study therefore was to investigate the expression profile and the role of PRDM genes in the molecular mechanism responsible of the proliferation-differentiation switch, paying particular focus on the PRDM2 gene expression in skeletal muscle C2C12 cell line, an established cell model for skeletal muscle differentiation studies [86,87], that has been successfully used to study differentiation and proliferation-differentiation switch [88].
Materials and Methods
Cell culture and transfection
C2C12 cells (kindly provided by Professor Fabio Naro from University “La Sapienza”-Rome) were maintained in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum, FBS (Invitrogen), referred as GM, i.e. growth medium, in humidified 95% air and 5% CO2. As the cells reached confluence, the medium was replaced with DMEM supplemented with 10 μg/ml insulin (Invitrogen). And 5 μg/ml transferrin (Invitrogen) referred as DM, i.e. differentiation medium, to induce differentiation of myoblasts into myotubes. Cell transfection with plasmid DNA was performed using Lipofectamine® 2000 Reagent (GIBCO BRL, Life Technologies, Rockville, MD, USA) in OptiMem I Reduced Serum Medium (Invitrogen) for 6 hours, according to the manufacturer’s instructions. Transfection medium was then removed, and cells were grown in DMEM supplemented with 5% FBS or DM, as indicated in the legend to figures, for additional 24-48-72 hours. Reverse transcriptase-polymerase chain reaction (RT-PCR)-Total RNA (1 µg) was extracted from C2C12 cells using Trizol reagent (Invitrogen), according to the manufacturer’s instructions. Gel electrophoresis in denaturing conditions was performed to evaluate the integrity of extracted RNA; the quality of RNA extracted was evaluated by the measure of 260/280 nm and 260/230 nm absorbance ratios (the threshold acceptance was 1.9 for absorbance ratio 260/280 nm and 2.2 for absorbance ratio 260/230). To remove contaminant DNA, RNA samples were treated with 40 U of RNase-free DNase-I (Boehringer Mannheim, Indianapolis, IN, USA) for 45 minutes at 37°C. The absence of contaminant genomic/plasmid DNA was checked by PCR of not reverse transcribed RNA samples. Total RNA was reverse transcribed using cDNA Synthesis Kit Transcriptor High Fidelity (Roche, Basilea, Switzerland). The cDNAs amplifications were performed by RT-PCR with specific primers set for glyceraldehyde-3-phosphate dehydrogenase (GAPDH)s, for MRF and for the detection of the PRDM plus transcript (PRDM PR as reported in table 1) and also for all PRDM transcripts (PRDM TOT as reported in table 1) using JF buffer (30mM Tris base, 8mM HEPES base, 20mM K glutamate, 60mM NH4 acetate, 2mM DTT, 8% glycerol, 1.5mM MgCl2, 0.2mM dNTPs) [79]. The reaction was performed using a thermal cycler (Eppendorf, Milan, Italy). Analysis of amplified products was done by electrophoresis on 2% agarose gel. The gel images were acquired by the Gel DOC XR System platform (Bio-Rad laboratories, Hercules, CA).
| Table 1: Sequences of primers used for RT-PCR analysis ZA | |
| PRDM2 PR F | ACTGGCTCCGCTATGTGAAC |
| PRDM2 PR R | CGCGATTGGCTTTAAGGTT |
| PRDM2 TOT F | CCGGAGAGGGAAGAAGAAAT |
| PRDM2 TOT R | TCATGTTTGCAGAGGTGGAG |
| PRDM10 PR R | TCACGAACTCAGCGTAGGATG |
| PRDM10 PR F | TGGTCCTCTACATAGATAGGTT |
| PRDM10 TOT F | TGAATGGACTAGATCAGCCAG |
| PRDM10 TOT R | AGCCCTTGTTACAGAGATCAC |
| PRDM4 PR F | AGCAGCTTGTTCTCCGCCAGTCC |
| PRDM4 PR R | GCACTCCTTGCCACAGTTACAGAG |
| PRDM4/PFM F | TCTCCCTCTCACAGTGCCAT |
| PRDM4/PFM R | GATCTAGTGCTGAAGGGTTGTTGG |
| PRDM1 PR F | GACGGGGGTACTTCTGTTCA |
| PRDM1 PR R | GGCATTCTTGGGAACTGTGT |
| PRDM1 TOT F | ACACCGGGACTCCTACTCCT |
| PRDM1 TOT R | GTACGGTGGCAACAGGAACT |
| PRDM5 PR F | GAGCCGAGCTCATTTCCTC |
| PRDM5 PR R | GTACGTACATGCCCAGCATC |
| PRDM5 TOT F | GCCATTCAAACACACACAGG |
| PRDM5 TOT R | TTTCACAGTACGGGCATTGA |
| PRDM14 PR F | TTGGTGATGTGCCACACTTT |
| PRDM14 PR R | TCCAGTTCCCAGAACCTTTG |
| PRDM14 TOT F | CACTCCCGAAGTACCACGAT |
| PRDM14 TOT R | CCCACCTCTGACCACTGATT |
| PRDM16 PR F | TTCCAATCCCACCAGACTTC |
| PRDM16 PR R | CATCTGCTCCCATCCAAAGT |
| PRDM16 TOT F | TGGGCTCACTACCCTACCAC |
| PRDM16 TOT R | GACTTTGGCTCAGCCTTGAC |
| MyoD F | TGGGATATGGAGCTTCTATCGC |
| MyoD R | GGTGAGTCGAAACACGGATCAT |
| Myf-5 F | TGAAGGATGGACATGACGGACG |
| Myf-5 R | TTGTGTGCTCCGAAGGCTGCTA |
| Myogenin F | AGGAGAGAAAGATGGAGTCCAGAG |
| Myogenin R | TAACAAAAGAAGTCACCCCAAGAG |
| c-Myc F | ATGCCCCTCAACGTGAACTTC |
| c-Myc R | CGCAACATAGGATGGAGAGCA |
| GAPDH F | TGTGTCCGTCGTGGATCTGA |
| GAPDH R | CCTGCTTCACCACCTTCTTGA |
Quantitative Reverse-Transcription PCR (qRT-PCR) Analysis-Aliquots of cDNA were subjected to quantitative analysis by real-time PCR (qRT-PCR) using the SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) in a Mastercycler ep Realplex (Eppendorf), as previously described [89]. The amplification was performed using the same primer sets of semi-quantitative RT-PCR. GAPDH was used as housekeeping gene for normalization. The dissociation curves showed a single peak and the agarose gel analysis of PCR amplicons showed a single DNA band of the expected molecular size. The relative quantification (RQ) was performed using the Δ-ΔCt method [90]. Data were presented as RQ (Relative Quantification) by comparing the threshold cycle of PCR products to the threshold cycle of standard cDNA. The linearity and the efficiency of the PCR reaction were analyzed by serial cDNA dilutions. Each amplification was done in triplicate and the reaction specificity was assessed by melting curves analysis. PRDM2 PR silencing by RNA interference -The gene silencing of PRDM2 PR gene product was achieved using the BLOCK-iT™ Pol II miR RNAi with emGFP expression vector (Invitrogen), according to the manufacturer’s instructions. The oligonucleotides, Hmi418948 and Hmi418951, which target PRDM2 PR gene product (Invitrogen Co.), were cloned into the vector (pcDNA™ 6.2-GW/EmGFP-miR). C2C12 cells were transfected with recombinant vector or with kit control vector (β-galactosidase, CTRL). Protein assay -The protein concentration was measured with BIO-RAD Protein Assay (Bio-Rad Laboratories, Hercules, CA) according to the manufacturer’s instructions.
Western blot analysis- C2C12 cells were washed with PBS and lysed in RIPA buffer 1X [50 mM Tris HCl pH 7.5, 150 mM NaCl, 0.25 % sodium deoxycholate, 1% NP-40 supplemented with protease inhibitor cocktail (Roche, Basilea, Svizzera). The clarified lysates were processed for Sodium Dodecyl Sulphate - PolyAcrylamide Gel Electrophoresis, SDS- PAGE (10% polyacrylamide gel) according to Laemmli procedure [91]; subsequently the proteins were transferred to PVDF membrane (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) using transfer buffer. Blots were blocked with 20 mM Tris pH 7.8, 100 mM NaCl, 0.1% Nonidet P40/Tween 20(1:1), 5% non-fat milk and incubated with primary antibodies diluted in the same buffer (0.1ml/cm2). The following antibodies were used at the indicated concentrations: mouse monoclonal anti-MyoD: sc-32758 (Santa Cruz Biotechnology) at the following concentration 0.2μg/ml; mouse monoclonal anti- Myc: sc-40 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) at the following dilution 1:500; mouse monoclonal anti-Myogenin: sc12732 (Santa Cruz Biotechnology, Inc.,Santa Cruz,CA) at the following dilution 1:500; rabbit polyclonal antibodies to RIZ1, RIZ 9710 (Abcam Ltd., Cambridge, UK) at the following dilution 1:1000, rabbit polyclonal antibodies against PRDM4 and PRDM10 (EPIGENTEK) at the following dilution 1:1000. Thereafter the same filter was stripped and reprobed with mouse monoclonal antibody against Histone H1 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) at the following dilution 1:1000. Peroxidase-conjugated anti-rabbit IgG or anti-mouse IgG secondary antibodies were used at 1:5000 dilution. Peroxidase activity was detected using an Amersham ECLTM Advance Western Blotting Detection Kit (GE Healthcare, Little Chalfold, UK), according to the manufacturer’s instructions. Membranes were exposed with Amersham Hyperfilm ECL film (GE Healthcare, Little Chalfold, UK) and images were acquired with the Gel DOC XR Platform (Bio-Rad Laboratories, Hercules, CA).
Cell growth analysis- Cell proliferation was evaluated by cell counting, by MTT (3-(4,5dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay (Sigma-Aldrich, Co. St. Louis, MO, USA) and by FACS (Fluorescence Adsorbed Cell Sorting, Becton Dickinson, New Jersey, USA) analysis as previously indicated [58].
Immunofluorescence analysis
Myoblasts were plated on poly-L-lysine (3x)-coated coverslip and cultured in DM for 72h. Subsequently DM was discarded and cells washed three times with PBS and incubated at room temperature with freshly-made paraformaldehyde (4% w/v) for 10 min. The cells were washed with glycine 0.1% for 5 min, followed by three washing in PBS, subsequently incubated for 2 hours with the following primary antibodies: (a) mouse monoclonal anti-MyHC (Sigma-Aldrich, Co. St.Louis, MO,USA) at the following dilution 1:500; (b) RIZ 9710 (Abcam Ltd, Cambridge, UK) at the following dilution 1:100. The primary antibodies were diluted in PBS containing 10% (v/v) fetal bovine serum (FBS) and 0.1% Triton X-100. Cells were then washed in PBS and incubated at room temperature for 1 h with fluorescent secondary antibodies (Jackson Laboratories, USA): (a) anti-mouse or anti-rabbit IgG conjugated to Texas red were diluted 1:100 in PBS containing 10%(v/v) fetal bovine serum (FBS) and 0.1% Triton X-100. Subsequently, coverslips were placed onto untreated glass slides and allowed to air dry. Coverslips were analyzed using a Zeiss LSM 510 Meta argon/krypton laser scanning confocal microscope. Each image was acquired 4 times and the signal averaged to improve the signal to noise ratio. Computer-assisted quantitative evaluation of channel distribution in cell compartment was done using the Image-J software.
Chromatin immunoprecipitation (ChIP)
C2C12 cells (approximately 5x106 cells/plate) were cultured in GM or in DM for 24, 48 and 72 hours. One-tenth aliquots were immunoprecipitated using the following antibodies: 1µg of purified IgG control antibody (Sigma-Aldrich), 1mg of rabbit polyclonal anti-RIZ1 (Abcam Ltd, Cambridge, UK) or 1 µg of mouse monoclonal anti-MyoD (Santa Cruz Biotechnology Inc., Santa Cruz, CA). Secondary immunoprecipitation was performed with Sepharose coupled to protein A (Sigma-Aldrich, Co. St. Louis, MO, USA). One-twenty-fifth of the DNA extracted from each immunoprecipitation was amplified using primers complementary to the Myogenin promoter region E2_E1 Forward-GAATCACATGTAATCCACTGGA E2_E1 Reverse- ACGCCAACTGCTGGGTGCCA). Amplifications were performed for an empirically determined number of PCR cycles producing a linear correlation between amplified band signals and template dilutions. AmpliTaq polymerase in AB1 buffer (30mM Tris base, 10mM HEPES base, 25mM KCl, 20mM K glutamate, 20mM NH4 acetate, 1.25mM DTT, 5% glycerol, 1.25mM MgCl2, 0.2mM dNTP) (Applied Biosystems, Foster City, CA) was used for all PCR analyses. Amplification products were analyzed by 2% agarose gel electrophoresis and bands were visualized with the Gel DOC XR System (Bio-Rad laboratories, Hercules, CA); densitometric analysis was performed using Total Lab 1D software. Statistical analysis-Statistical significance was determined with a paired t-test with Graphpad prism 5.0 software (La Jolla, CA, USA).
Results
Analysis of the expression levels of MRF genes and PRDM genes in myoblasts and myotubes
To induce differentiation of myoblasts into myotubes, confluent C2C12 cells were incubated in low mitogenic media (DM) and after 72 hours, differentiation is complete as observed by morphological changes. To verify the fidelity of the model used, an immunofluorescence analysis (Figure 1S a) of C2C12 cells differentiated in the same experiment was performed. The results revealed that after 72 h in DM medium cells formed myotubes expressing the myosin heavy chain (MyHC), a marker of myoblasts differentiation (Figure 1S b-c) [92]. The expression level of PRDM genes (PRDM1, 2, 4, 5, 10, 14 and 16) and MRF genes: Myc and Myogenin was evaluated by qRT-PCR analysis. The amplification of PR plus product was performed with a set of primer amplifying a transcript region encoding for the PR domain (PRDM PR F/R) and another primer set was used for amplification of a region common to both PR plus and PR minus forms (PRDMtot F/R). As expected, the low mitogenic environment increased the expression level of differentiation markers Myogenin and reduced the proliferation markers c-Myc (Figure 1). In our experimental conditions, myotubes showed no significant variation in the PRDM1, PRDM5, PRDM14 and PRDM16 expression level than myoblasts. On the other hand, PRDM2, PRDM4 and PRDM10 expression levels falls down into myotubes than myoblasts. So we decided to further investigate the variation in the expression level of PRDM genes that showed a significantly difference in the expression level between myoblasts and myotubes.
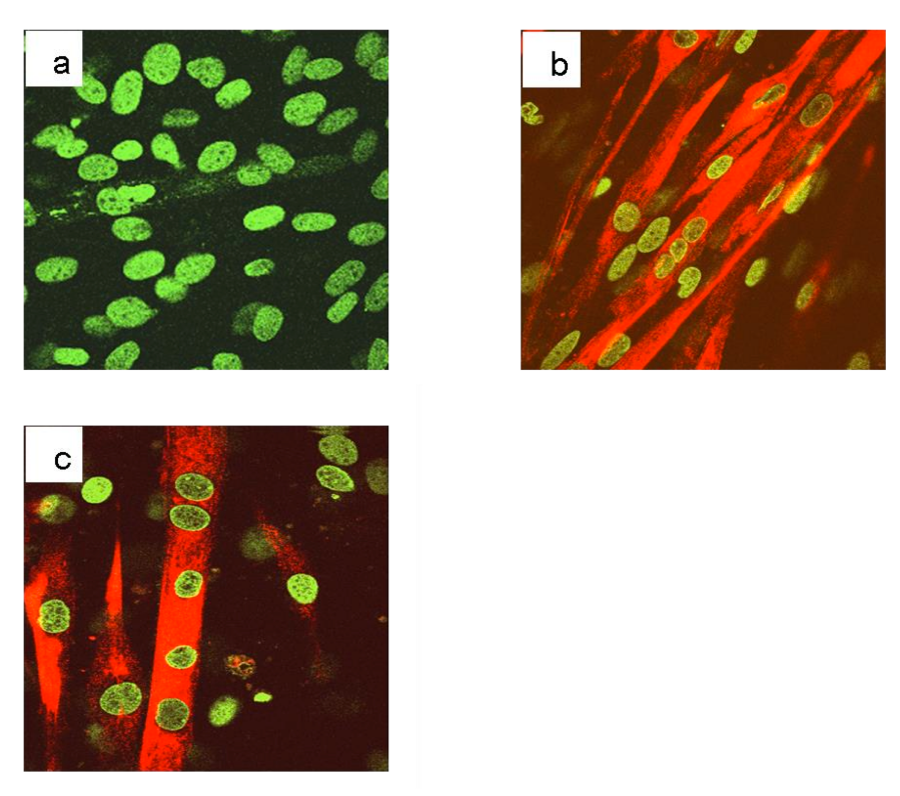
Figure 1S: Immunofluorescence analysis of the differentiation marker, myosin heavy chain, MyHC, in C2C12 cell line cultured in GM or DM. C2C12 cells were plated on glass slides for immunofluorescence and cultured for additional 120 hours in DM. After treatment, the cells were fixed, permeabilized and incubated with an antibody against the differentiation marker, myosin heavy chain, MyHC. Nuclei were stained with the chromomycin dye. The slides were analyzed by confocal fluorescence microscopy and significant images were acquired and displayed.
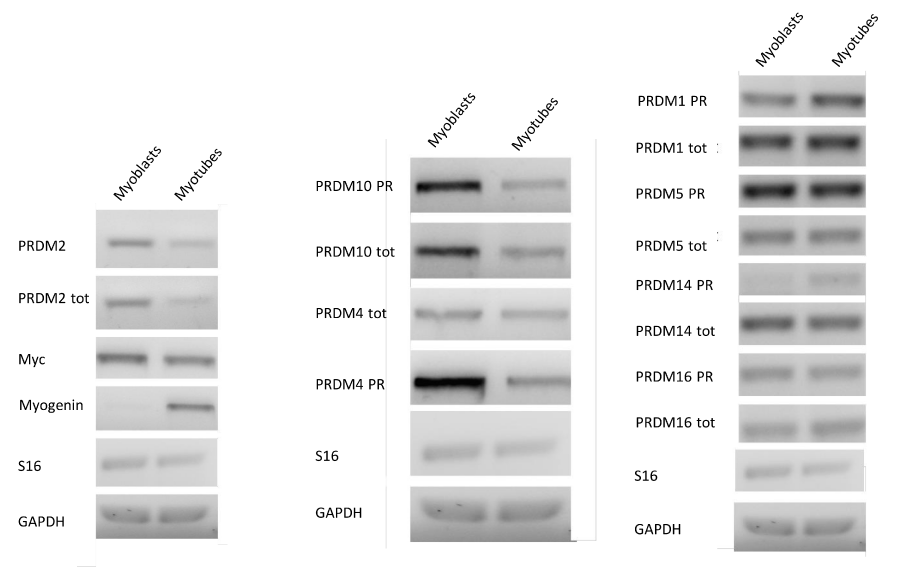
Figure 1: MRF and PRDM gene expression in myoblasts and myotubes. The electrophoretic analysis shows the fragments obtained by RT-PCR of total mRNA/cDNA extracted from C2C12 cell. The amplification of PR plus product was performed with a set of primer amplifying a transcript region encoding for the PR domain (PRDM PR F/R) and another primer set was used for amplification of a region common to both PR plus and PR minus forms (PRDMtot F/R).
Analysis of the expression levels of MRF genes and PRDM genes during differentiation of myoblasts in myotubes
To induce differentiation of myoblasts into myocytes, confluent C2C12 cells were incubated in low mitogenic media (DM) and at different time (0, 24, 48, 72 hours) was evaluated by qRT-PCR the expression level of MRF genes: Myc, Myf5, MyoD and Myogenin. As expected, the low mitogenic environment increased quickly the expression of differentiation markers MyoD and Myogenin and reduced the proliferation markers Myc and Myf5 expression level (Figure 2). It has been extensively demonstrated by Cheedipudi et al. [87], that PRDM2 gene expression levels is lower in myotubes than myocytes but no evidence are available on the variation of other member of PRDM family gene expression level during differentiation induction. In order to determine the precise temporal gap in which occurred PRDM expression level variation, qRT-PCR analysis of total C2C12 RNA extracted at different time (24, 48 and 72 hours) from differentiation induction was performed. As shown in figure 3A and B, at 24 h from differentiation induction PRDM2, PRDM4 and PRDM10 genes were expressed at lower level then myoblasts. This reduction was more evident at 72 h for PRDM2 PR plus product. Interestingly and according to Cheedipudi [87], the maximal expression of Myogenin at 72 h from differentiation induction corresponds to the reduction of band intensity of PRDM2 PR. The particular behaviour of PRDM2 gene prompted us to investigate the relationship between PRDM2 gene and myoblasts differentiation. Immunofluorescence analysis (Figure 2S) of C2C12 in GM or after culture in DM for 24 h revealed that RIZ1 in proliferating condition is predominantly localized in the nucleus and translocates into the cytoplasm when C2C12 cells are committed to differentiate.
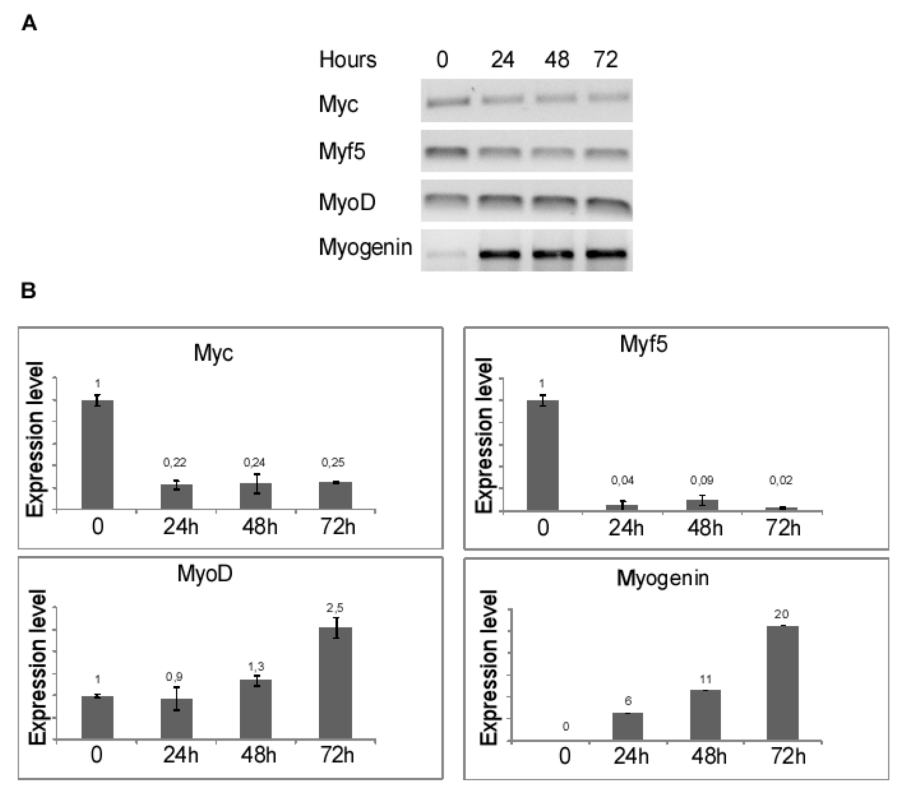
Figure 2: MRF genes expression after induction of differentiation. The transcripts encoded by MRF genes was measured by RT-PCR and qRT-PCR after 24, 48, 72 hours of differentiation induction (see Materials and methods section). The expression level is indicated as fold changes from basal conditions. A) Electrophoretic analysis of the fragments obtained by RTPCR of total mRNA/cDNA extracted from C2C12 cells; B) qRT-PCR of mRNA/cDNA extracted from C2C12 cells. Histograms represent the averages (+/− standard error) from at least three independent experiments, normalized for the expression of the control housekeeping gene GAPDH, reported in figure 2 (# indicates p < 0.05 for each gene versus control).
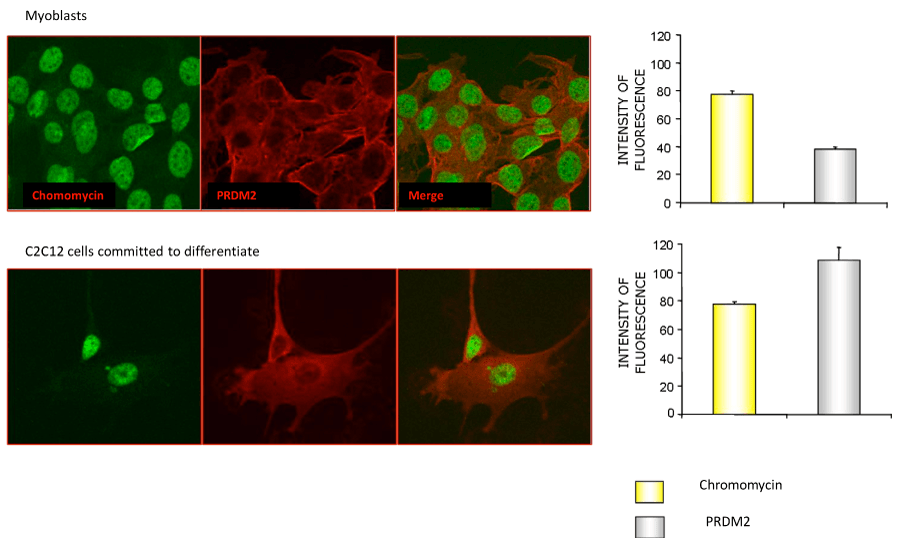
Figure 2S: Immunofluorescence analysis of the RIZ1 protein in the C2C12 cell line cultured in GM or DM. C2C12 cells were plated on glass slides for immunofluorescence and cultured for additional 24 hours in GM or DM. After treatment, the cells were fixed, permeabilized and incubated with an antibody against the RIZ1 protein. Nuclei were stained with the chromomycin dye. The slides were analyzed by confocal fluorescence microscopy and significant images were acquired and displayed.
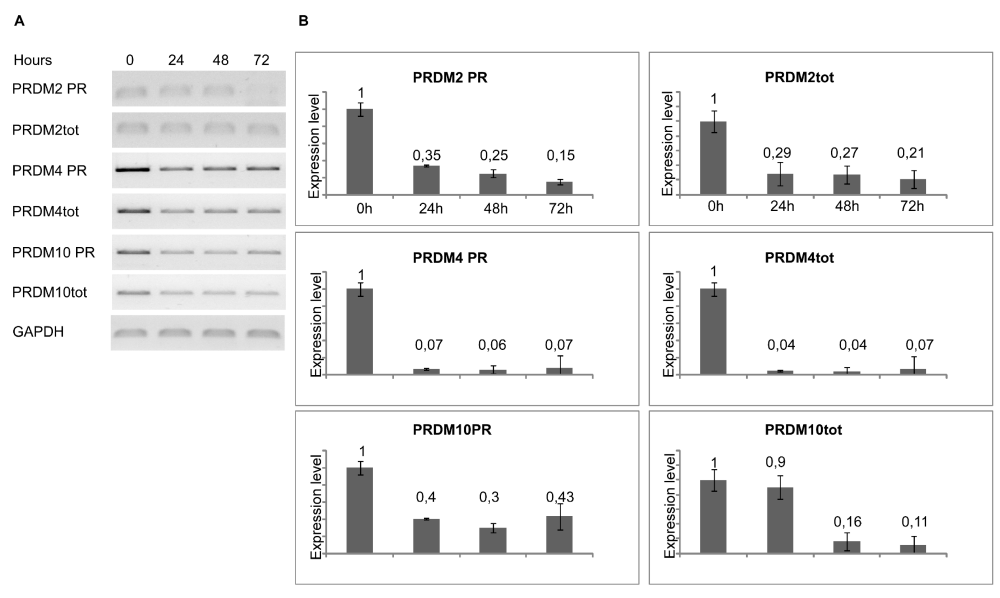
Figure 3: Modulation of PRDM genes expression during differentiation. The transcripts encoded by PRDM genes was measured by RT-PCR and qRT-PCR after 24, 48, 72 hours of differentiation induction (see Materials and methods section). The expression level is indicated as fold changes from basal conditions. The PRDM PR and PRDM2tot sets of primers recognize sequences on the region coding PR domain of PRDM genes or on a region common to both PRDM PR plus and minus, respectively. A) Electrophoretic analysis of the fragments obtained by RT-PCR of total mRNA/cDNA extracted from C2C12 cells; B) qRT- PCR of total mRNA/cDNA extracted from C2C12 cells. Histograms represent the averages (+/− standard error) from at least three independent experiments, normalized for the expression of the control housekeeping gene GAPDH (# indicates p < 0.05 for each form of PRDM genes versus their untreated control).
Effect of PRDM2 silencing on C2C12 myoblasts differentiation
As observed by qRT-PCR analysis of PRDM2/RIZ1 (60%) forced suppression with the plasmid coding the miRNAPRDM2 spanning a sequence coding for the aa 97-103 of RIZ1 protein, induced an increase in expression level of the differentiation markers MyoD and Myogenin that was more evident for the last one (Figure 4). This evidence confirms that RIZ1 protein negatively controls the induction of differentiation mediated by MRF factor. Western blot analysis of whole cell lysates from C2C12 transiently transfected with a control plasmid or with the plasmid coding the miRNAPRDM2, showed a strong reduction in the RIZ1 expression level, revealing the efficacy of the interference (Figure 5). The PRDM2/RIZ1 silencing induced a decrease in the PRDM4 and PRDM10 expression levels. These evidence support the hypothesis that different genes of PRDM family control proliferation/differentiation switch and are probably organized in a hierarchical manner.
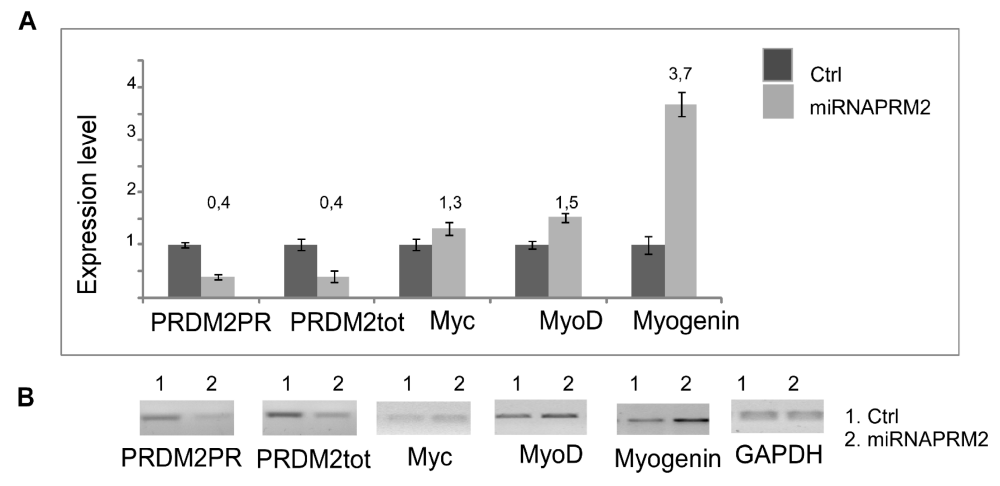
Figure 4: Modulation of MRF genes expression during differentiation. The transcripts encoded by PRDM genes was measured by RT-PCR and qRT-PCR after 24, 48, 72 hours of differentiation induction (see Materials and methods section). The expression level is indicated as fold changes from basal conditions. The PRDM PR and PRDM2tot sets of primers recognize sequences on the region coding PR domain of PRDM genes or on a region common to both PRDM PR plus and minus, respectively. A) Electrophoretic analysis of the fragments obtained by RT-PCR of total mRNA/cDNA extracted from C2C12 cells; B) qRTPCR of total mRNA/cDNA extracted from C2C12 cells. Histograms represent the averages (+/− standard error) from at least three independent experiments, normalized for the expression of the control housekeeping gene GAPDH (# indicates p < 0.05 for each form of PRDM genes versus their untreated control).
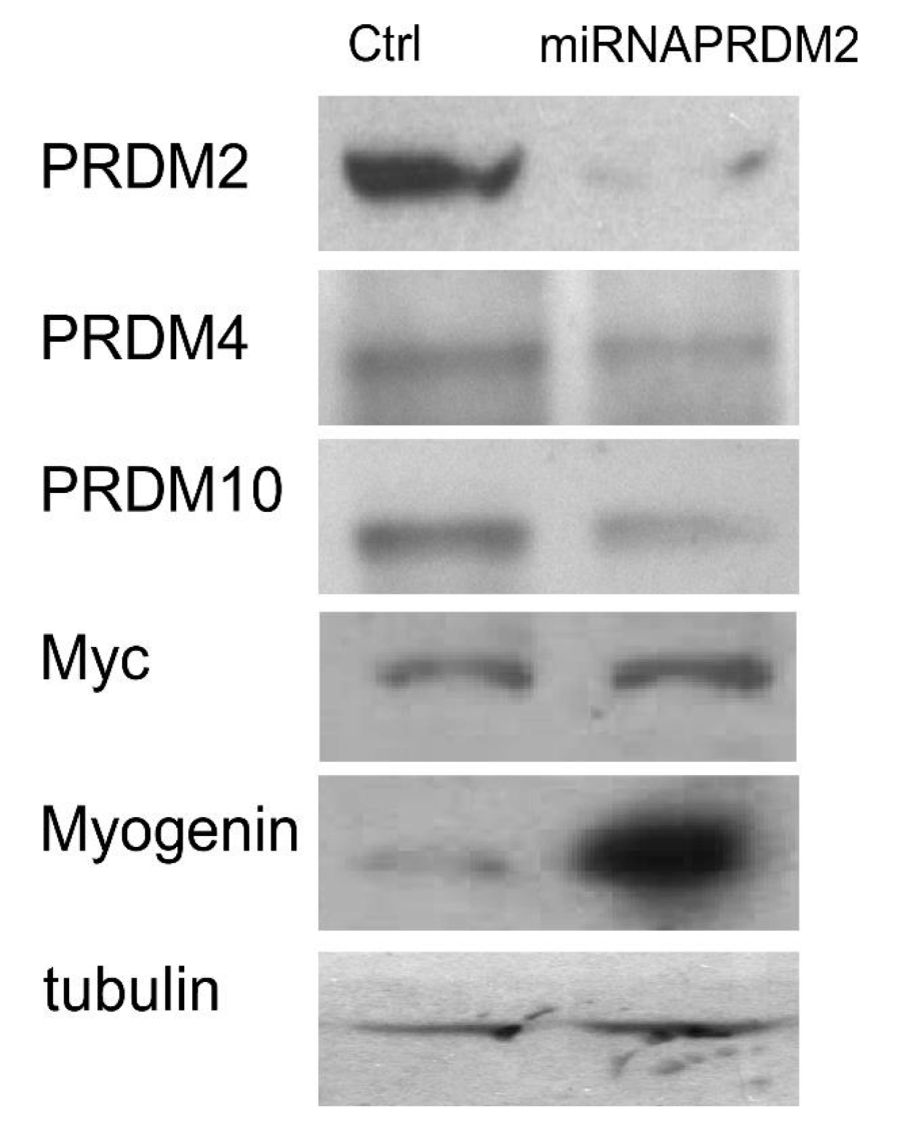
Figure 5: Modulation of PRDM and MRFs proteins expression into PRDM2 silenced C2C12 myoblasts. The expression level of protein encoded by PRDM2, PRDM4 and PRDM10 genes and MRFs was evaluated by SDS-PAGE and Western blot analysis of total protein cell extracts from C2C12 myoblasts and PRDM2 silenced C2C12 myoblasts (see Materials and methods section). The blots are representative of three independent experiments.
Interaction of RIZ with E2/E1 Myogenin promoter region
ChIP experiments were performed to detect the presence of MyoD, a master regulator of the skeletal muscle gene expression program [28], and RIZ on E2/E1 Myogenin promoter region during differentiation induction. Therefore, equivalent amounts of cross-linked chromatin from myoblasts cultured in GM or DM (24, 48 and 72 hours) were immunoprecipitated in parallel with two antibodies that recognize PRDM2/RIZ1 or MyoD. The precipitated DNA then was subjected to PCR amplification with the use of a primers set, spanning two of the E-box sites (E1 and E2) located in the proximal region of Myogenin promoter. Results shown in Fig. 6B, revealed that MyoD was ever associated with Myogenin promoter according to Mal et al., [93]; conversely, as shown in figure 6A, RIZ1 was associated with the E2/E1 Myogenin promoter region in myoblasts at 24h whereas at 48h and 72h no association of RIZ1 was detected. The specificity of this assay is demonstrated by the observation that the binding of MyoD or RIZ1 was not observed when a normal rabbit IgG was used in the analysis.
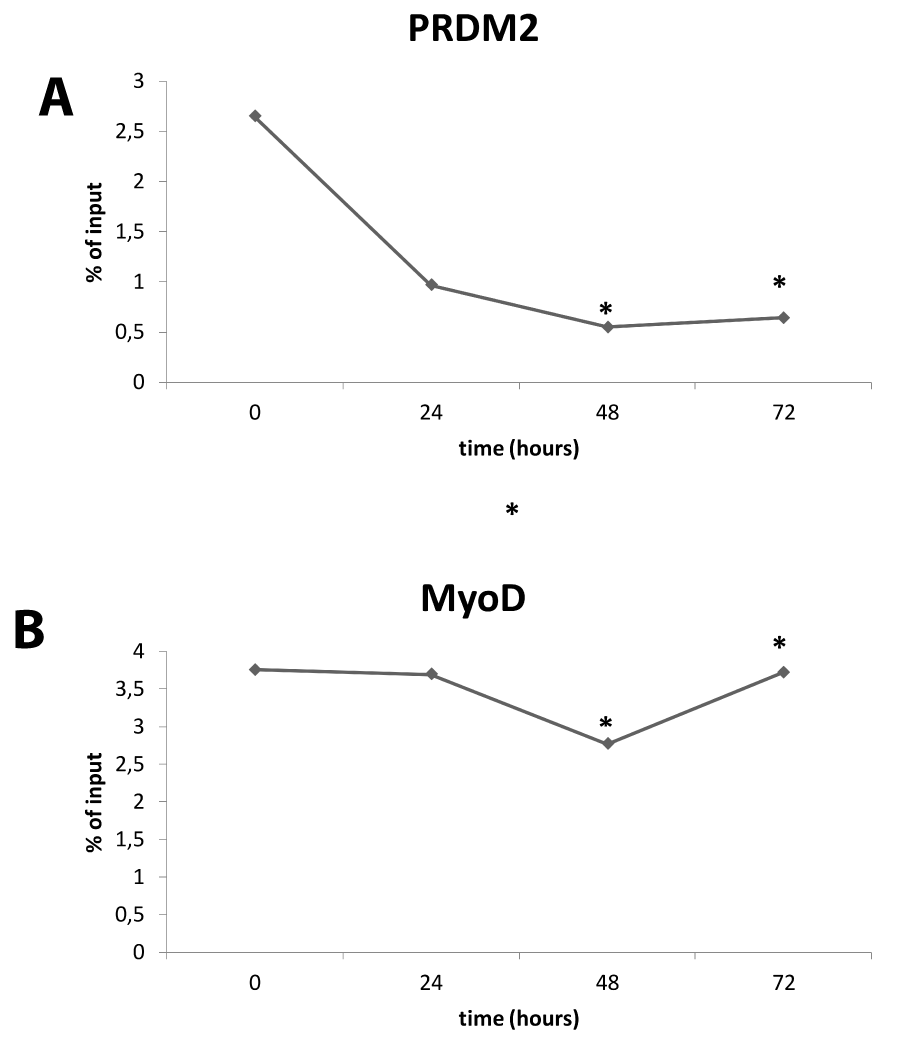
Figure 6: Interaction of RIZ with E1/E2 promoter. ChIP analysis of E2/E1 myogenin promoter region. Densitometric analysis of bands amplified from ChIP samples with antibodies to RIZ1 (upper panel) or to MyoD (lower panel), expressed as percentage of the input after background subtraction. The graph is representative of three independent experiment (* indicates p < 0.05 vs control).
RIZ1 silencing induced cell cycle arrest
In order to investigate the effect of RIZ silencing on proliferation rate, we performed a cell cycle analysis evaluating propidium iodide incorporation by FACS. As shown in figure RIZ1 silencing by miRNA induced a G1 phase delay. Likewise MTT assays of C2C12 cells transfected with the miRNAPRDM2 plasmid significantly reduced the cells number (Figure 7).
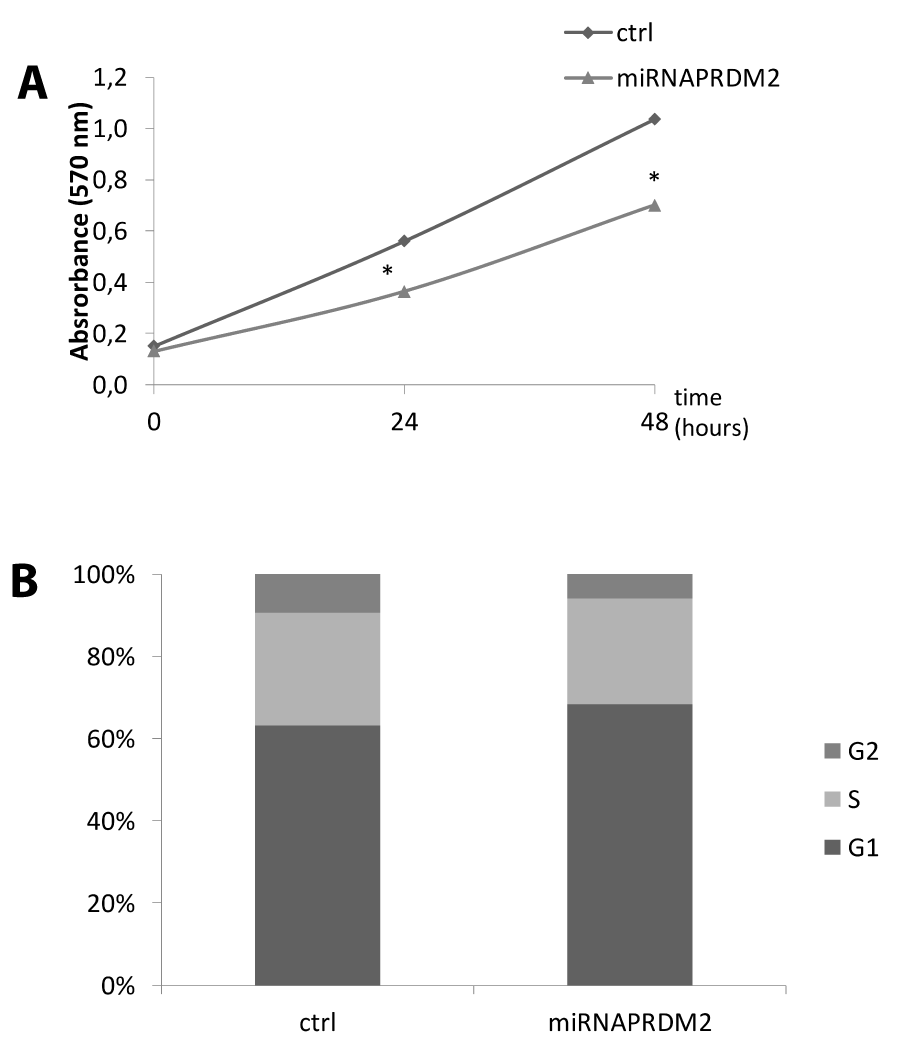
Figure 7: Effect on cell proliferation and survival upon RIZ1 knock down in C2C12 cell line. (A) For the colorimetric MTT assay, C2C12 cells transiently transfected with a plasmid encoding a miRNA against RIZ1 or a not-targeting plasmid (ctrl) were plated at the same density and allowed to grow for 0, 24 or 48 h. MTT was added in the last 2 h, formazan precipitates were dissolved with dimethyl sulfoxide reagent and absorbance read at 570 nm [31]. Values are the mean (±SE) of three analyses from three independent experiments # p < 0.05 vs. control; (B) C2C12 cells transiently transfected with a plasmid encoding a miRNA against RIZ1 or a not-targeting plasmid (ctrl) were cultured in 60-mm dishes for 48 h. Cells were processed following manufacturer’s instruction and finally analyzed by flow cytometer to determine the percentage of cells in the different phases of cell cycle. The data are the mean of three independent experiments performed in triplicate ( = 9) # p < 0.05 vs control.
Discussion
PRDM family gene plays a key role in the control of a plethora of cell life processes, like cell cycle progression, homeostasis maintenance of immune system and control of early stage of development. PRDM1 and PRDM14 for example guide the epigenetic reprogramming, necessary to determinate the progenitors of germ cells during embryonic development; PRDM9 also is indispensable for meiotic prophase progression during gametogenesis; PRDM16 controls the cell fate switch between myoblasts and brown adipocytes: recently studies, conducted by Li et colleagues, demonstrated that C2C12 myoblasts, stably transfected with PRDM16, showed a repression of myogenic genes and an upregulation of adipogenic genes at proliferation and differentiation genes, probably due to CpG methylation of MyoD [84]. To this behaviour, the study of molecular basis of PRDM action mechanism appears very important. Data obtained by RT-PCR and Western blot analysis in C2C12 cell line showed that the expression of PRDM2 gene was selectively modulated during differentiation, because of the differentiated cells showed reduced levels of expression of RIZ1 than myoblasts. This trend is in disagreement with the experimental evidence obtained in epithelial cell line models [78] and lymphocytes [94] in which treatment with differentiating agents, or the stimulation of cell proliferation, respectively, caused the increase and decreased of PRDM2/RIZ1 expression. On the contrary, there are some similarities with the experimental evidence obtained with the osteosarcoma cell line (SAOS2), in which PRDM2/RIZ1 showed an elevated expression in proliferating cells, compared to cells that had temporarily left the cell cycle [95]. These suggestions left us to speculate that RIZ can be related to the cell type and in the mesenchymal cells perhaps cell cycle escape and subsequent differentiation coincide with RIZ1 expression level reduction: immunocytochemistry experiments, performed in myoblasts and myotubes, showed that during differentiation there is an intracellular redistribution of RIZ protein, predominantly localized in the perinuclear and nuclear area. This phenomenon, as myeloid leukemia cell line HL60, could be interpreted as an intranuclear confinement of RIZ protein. Expression level analysis by qRT-PCR of other member of PRDM family gene PRDM4 and PRDM10 showed that the differentiation induction was characterized by a reduction of its expression levels. In order to confirm the involvement of PRDM2 gene in the proliferation-differentiation switch, we performed knockdown experiments. Interestingly, the PRDM2 gene silencing was characterized by a Myogenin expression level increasing, suggesting its peculiar role in the transcriptional control of myogenin expression. ChIP experiments performed with MyoD and RIZ antibodies on E2/E1 Myogenin promoter region in proliferation condition and after 24 h from differentation induction, confirmed a binding on the Myogenin promoter. As demonstrated by Mal A. [93], Lys-9 of H3 histones were methylated by SUV39H1 [96], surrounding the myogenin promoter in undifferentiated myoblasts. This marking was dramatically reduced in myoblasts that had undergone differentiation. We hypothesize that another member of SET family protein, RIZ1, can modulate myogenin expression, and RIZ1 interference reduced the expression level of PRDM4 and PRDM10 genes, lead us to speculate that the members of the family have a redundant PRDM behavior and/or cooperate in the transition proliferation-differentiation. Futhermore PRDM2 silencing induced a G1 phase delay and a reduction of proliferation.
Conclusion
In conclusion, our studies suggest that PRDM2, PRDM4 and PRDM10 play a pivotal role in the proliferation-differentiation switch of myoblast in myotubes.
Acknowledgements
This work was supported by grants from the Italian Ministry of University and Scientific Research PRIN.
References
- Singh K, Dilworth FJ. Differential modulation ofcell cycle progression distinguishes members of the myogenic regulatoryfactor family of transcription factors. FEBS J. 2013; 280: 3991-4003. Ref.: https://goo.gl/mw5Y9p
- Davis RL, Weintraub H. Acquisition of myogenicspecificity by replacement of three amino acid residues from MyoD intoE12. Science. 1992; 256: 1027-1030. Ref.: https://goo.gl/kD7f3S
- Tapscott SJ, Davis RL, Thayer MJ, Cheng PF,Weintraub H, et al. MyoD1: a nuclear phosphoprotein requiring a Mychomology region to convert fibroblasts to myoblasts. Science. 1988; 242:405-11. Ref.: https://goo.gl/6eXUeW
- Atchley WR, Fitch WM, Bronner-Fraser M. Molecularevolution of the MyoD family of transcription factors. Proc Natl AcadSci.1994; 91: 11522-11526. Ref.: https://goo.gl/d2oZHf
- Berkes, CA, Tapscott SJ. MyoD and thetranscriptional control of myogenesis. Semin Cell Dev Biol. 2005; 16: 585-595.Ref.: https://goo.gl/cSHJLx
- Braun T, Buschhausen-Denker G, Bober E, TannichE, Arnold HH. A novel human muscle factor related to but distinct fromMyoD1 induces myogenic conversion in 10T1/2 fibroblasts. EMBO J. 1989; 8:701-709. Ref.: https://goo.gl/Vwy7uo
- Kablar B, Rudnicki MA. Development in the absenceof skeletal muscle results in the sequential ablation of motor neuronsfrom the spinal cord to the brain. Dev Biol. 1999; 208: 93-109. Ref.: https://goo.gl/z5nXSY
- Rudnicki, MA, Schnegelsberg PN, Stead RH, BraunT, Arnold HH., et al. MyoD or Myf-5 is required for the formation ofskeletal muscle. Cell 1993; 75: 1351-1359. Ref.: https://goo.gl/cWenqL
- Tajbakhsh S, Rocancourt D, Buckingham M. Muscleprogenitor cells failing to respond to positional cues adopt non-myogenicfates in myf-5 null mice. Nature. 1996; 384: 266-270. Ref.: https://goo.gl/FpK37B
- Reijntjes S, Francis-West P, Mankoo BS. Retinoicacid is both necessary for and inhibits myogenic commitment anddifferentiation in the chick limb. Int J Dev Biol. 2010; 54: 125-134. Ref.: https://goo.gl/pD5Xh2
- Ustanina S, Carvajal J, Rigby P, Braun T. Themyogenic factor Myf5 supports efficient skeletal muscle regeneration byenabling transient myoblast amplification. Stem Cells. 2007; 25: 2006-2016.Ref.: https://goo.gl/qJ7YU6
- Gayraud-Morel B, Chrétien F, Flamant P, Gomès D,Zammit PS, Tajbakhsh S. A role for the myogenic determination gene Myf5 inadult regenerative myogenesis. Dev Biol. 2007; 312: 13-28. Ref.: https://goo.gl/DR6jKq
- Montarras D, Lindon C, Pinset C, Domeyne P.Cultured myf5 null and myoD null muscle precursor cells display distinctgrowth defects. Biol cell. 2000; 92: 565-572. Ref.: https://goo.gl/rQLzi9
- Panda AC, Abdelmohsen K, Martindale JL, DiGermanio C, Yang X, et al. Novel RNA-binding activity of MYF5 enhancesCcnd1/Cyclin D1 mRNA translation during myogenesis. Nucleic Acids Res.2016; 44: 2393-2408. Ref.: https://goo.gl/oKMM6P
- Wright WE, Sassoon DA, Lin VK. Myogenin, a factorregulating myogenesis, has a domain homologous to MyoD. Cell. 1989; 56:607-617. Ref.: https://goo.gl/DmD2qg
- Hashimoto N, Ogashiwa M, Iwashita S. Role oftyrosine kinase in the regulation of myogenin expression. Eur J Biochem 1995;227: 379-387. Ref: https://goo.gl/Lz6TQF
- Rhodes SJ, Konieczny SF. Identification of MRF4:a new member of the muscle regulatory factor gene family. Genes Dev. 1989;3: 2050-2061. Ref: https://goo.gl/uMgCbh
- Arnold HH, Braun T. Targeted inactivation ofmyogenic factor genes reveals their role during mouse myogenesis: areview. Int J Dev Biol. 1996; 40: 345-353. Ref: https://goo.gl/WJ5MPb
- Moncaut N, Rigby PWJ, Carvajal, JJ. Dial M (RF)for myogenesis. FEBS J. 2013; 280: 3980-3990. Ref: https://goo.gl/YSZk62
- Chandra S, Terragni J, Zhang G, Pradhan S,Haushka S, et al. Tissue-specific epigenetics in gene neighborhoods:myogenic transcription factor genes. Hum Mol Genet. 2015; 24: 4660-4673. Ref: https://goo.gl/4SPWMT
- Molkentin JD, Black BL, Martin JF, Olson EN.Cooperative activation of muscle gene expression by MEF2 and myogenic bHLHproteins. Cell. 1995; 83: 1125-1136. Ref: https://goo.gl/YLEyhp
- Pon JR, Marra MA. MEF2 transcription factors:developmental regulators and emerging cancer genes. Oncotarget. 2016; 7:22970-22312. Ref: https://goo.gl/kbzRUj
- Cao Y, Yao Z, Sarkar D, Lawrence M, Sanchez GJ, etal. Genome-wide MyoD binding in skeletal muscle cells: a potential forbroad cellular reprogramming. Dev Cell 2010; 18: 662-674. Ref: https://goo.gl/M39LeS
- Soleimani VD, Yin H, Jahani-Asl A, Ming H, KockxCEM, et al. Snail regulates MyoD binding-site occupancy to direct enhancerswitching and differentiation-specific transcription in myogenesis. MolCell. 2012; 47: 457-468. Ref: https://goo.gl/HTLeyZ
- Murre C, McCaw PS, Vaessin H, Caudy M, Jan LY, etal. Interactions between heterologous helix-loophelix proteins generatecomplexes that bind specifically to a common DNA sequence. Cell 1989; 58:537-544. Ref: https://goo.gl/vcQBSt
- Blackwell TK, Weintraub H. Differences andsimilarities in DNA-binding preferences of MyoD and E2A protein complexesrevealed by binding site selection. Science. 1990; 250: 1104-1110. Ref: https://goo.gl/MEyBj2
- Blais A, Tsikitis M, Acosta-Alvear D, Sharan R,Kluger Y, et al. An initial blueprint for myogenic differentiation. GenesDev. 2005; 19: 553-569. Ref: https://goo.gl/FPp6dQ
- Aziz A, Liu QC, Dilworth FJ. Regulating a masterregulator: establishing tissuespecific gene expression in skeletal muscle.Epigenetics 2010; 5: 691-695. Ref: https://goo.gl/j6xEnj
- De Falco G, Comes F, Simone C. pRb: master ofdifferentiation. Coupling irreversible cell cycle withdrawal withinduction of muscle-specific transcription. Oncogene. 2006; 25: 5244-5249.Ref: https://goo.gl/ZPQ9W8
- Giacinti C, Bagella L, Puri PL, Giordano A,Simone C. MyoD recruits the cdk9/cyclin T2 complex on myogenic-genesregulatory regions. J Cell Physiol. 2006; 206: 807-813. Ref: https://goo.gl/mCAE7b
- Martelli F, Cenciarelli C, Santarelli G, PolikarB, Felsani A, et al. MyoD induces retinoblastoma gene expression duringmyogenic differentiation. Oncogene. 1994; 9: 3579-3590. Ref: https://goo.gl/LVfg3U
- Halevy O, Novitch BG, Spicer DB, Skapek SX, RheeJ, et al. Correlation of terminal cell cycle arrest of skeletal musclewith induction of p21 by MyoD. Science. 1995; 267: 1018-1021. Ref: https://goo.gl/aXwh2j
- Basile V, Baruffaldi F, Dolfini D, Belluti S,Benatti P, et al. NF-YA splice variants have different roles on muscledifferentiation. Biochim. Biophys Acta 2016; 1859: 627-638. Ref: https://goo.gl/RXTp41
- Besson A, Dowdy SF, Roberts JM. CDK Inhibitors:Cell Cycle Regulators and Beyond. Dev Cell 2008; 14: 159-169. Ref: https://goo.gl/LYEJ4R
- Zhang P, Wong C, Liu D, Finegold M, Harper JW, etal. p21(CIP1) and p57(KIP2) control muscle differentiation at the myogeninstep. Genes Dev. 1999; 13: 213-224. Ref: https://goo.gl/V2osBr
- FumasoniI, Meani N, Rambaldi D, Scafetta G, Alcalay M, et al. Family expansion andgene rearrangements contributed to the functional specialization of PRDMgenes in vertebrates. BMC Evol Biol. 2007; 7: 187. Ref: https://goo.gl/w36vh7
- Vervoort M, Meulemeester D, Béhague J, Kerner P.Evolution of Prdm Genes in Animals: Insights from Comparative Genomics. MolBiol Evol. 2016; 33: 679-696. Ref: https://goo.gl/hQNY1L
- Kinameri E, InoueT, Aruga J, Imayoshi I, Kageyama R, et al. Prdm proto-oncogenetranscription factor family expression and interaction with the Notch-Hespathway in mouse neurogenesis. PLoS One. 2008; 3: e3859. Ref: https://goo.gl/26UZEg
- Huang S. Histone methyltransferases, dietnutrients and tumour suppressors. Nat Rev Cancer. 2002; 2: 469-476. Ref: https://goo.gl/b73pB9
- Fog CK, Galli GG, Lund AH. PRDM proteins:important players in differentiation and disease. Bioessays. 2012; 34: 50-60.Ref: https://goo.gl/G8bjFM Gyory I, Wu J, Fejér G, Seto E, Wright KL. PRDI-BF1recruits the histone H3 methyltransferase G9a in transcriptionalsilencing. Nat Immunol. 2004; 5: 299-308. Ref: https://goo.gl/z2qBqf
- Duan Z, Person RE, Lee HH, Huang S, Donadieu J,et al. Epigenetic regulation of protein-coding and microRNA genes by theGfi1-interacting tumor suppressor PRDM5. Mol Cell Biol. 2007; 27: 6889-6902.Ref: https://goo.gl/o9rsFs
- Davis CA, Haberland M, Arnold MA, Sutherland LB,McDonald OG, et al. PRISM/PRDM6, a transcriptional repressor that promotesthe proliferative gene program in smooth muscle cells. Mol Cell Biol. 2006;26: 2626-2636. Ref: https://goo.gl/F2qSto
- Zhang Y, Stehling-Sun S, Lezon-Geyda K, Juneja SC,Coillard L, et al. PR-domain-containing Mds1-Evi1 is critical for long-termhematopoietic stem cell function. Blood 2011; 118: 3853-3861. Ref: https://goo.gl/CAgCMP
- Derunes C, Briknarová K, Geng L, Li S, GessnerCR, et al. Characterization of the PR domain of RIZ1 histonemethyltransferase. Biochem Biophys Res Commun. 2005: 333: 925-934. Ref: https://goo.gl/6EFkQu
- Eom GH, Kim K, Kim SM, Kee H J, Kim JY, at al..Histone methyltransferase PRDM8 regulates mouse testis steroidogenesis. BiochemBiophys Res Commun 2009; 388: 131-136. Ref: https://goo.gl/F8KSX8
- Hayashi K, Yoshida K, Matsui YA. Histone H3methyltransferase controls epigenetic events required for meiotic prophase.Nature. 2005; 438: 374-378. Ref: https://goo.gl/eFVaU5
- Fears S, Mathieu C, Zeleznik-Le N, Huang S,Rowley JD, et al. Intergenic splicing of MDS1 and EVI1 occurs in normaltissues as well as in myeloid leukemia and produces a new member of the PRdomain family. Proc Natl Acad Sci USA. 1996; 93: 1642-1647. Ref: https://goo.gl/M8fJtd
- Sun X.J, Xu PF, Zhou T, Hu M, Fu CT, et al.Genome-wide survey and developmental expression mapping of zebrafish SETdomain-containing genes. PLoS One. 2008; 3: e1499. Ref: https://goo.gl/gcTckQ
- Wu H, Min J, Lunin VV, Antoshenko T, DombrovskiL, et al. Structural Biology of Human H3K9 Methyltransferases. PLoS One.2010; 5: e8570. Ref: https://goo.gl/Vy9nKX
- Zannino DA, Sagerström CG. An emerging role forprdm family genes in dorsoventral patterning of the vertebrate nervoussystem. Neural Dev. 2015; 10: 24. Ref: https://goo.gl/u2LDuH
- Rea S, Eisenhaber F, Carroll D, Strahl BD, Sun ZW,et al. Regulation of chromatin structure by site-specific histone H3methyltransferases. Nature. 2000; 406: 593-599. Ref: https://goo.gl/Pck8uA
- Hohenauer T, Moore AW. The Prdm family: expandingroles in stem cells and development. Development. 2012; 139: 2267-2282. Ref: https://goo.gl/jPCxk1
- Györy I, Fejér G, Ghosh N, Seto E, Wright KL.Identification of a functionally impaired positive regulatory domain Ibinding factor 1 transcription repressor in myeloma cell lines J Immunol.2003; 170: 3125-3133. Ref: https://goo.gl/rQW9ti
- Hirai H. The transcription factor Evi-1. Int JBiochem Cell Biol. 1999; 31: 1367-1371. Ref: https://goo.gl/3TKze6
- Liu L, Shao G, Steele-Perkins G, Huang S. Theretinoblastoma interacting zinc finger gene RIZ produces a PR domain-lackingproduct through an internal promoter. J Biol Chem. 1997; 272: 2984-2991. Ref: https://goo.gl/A3RY5D
- Zazzo E, De Rosa C, Abbondanza C, Moncharmont. BPRDM Proteins: Molecular Mechanisms in Signal Transduction andTranscriptional Regulation. Biology (Basel). 2013; 2: 107-141. Ref: https://goo.gl/GA4nXP
- Zazzo E, Porcile C, Bartollino S, Moncharmont B.Critical Function of PRDM2 in the Neoplastic Growth of Testicular Germ CellTumors. Biology (Basel). 2016; 5: 54. Ref: https://goo.gl/NAKVav
- Geli J, Kiss N, Kogner P, Larsson C. Suppressionof RIZ in biologically unfavourable neuroblastomas. Int J Oncol. 2010; 37:1323-1330. Ref: https://goo.gl/CGkbaM
- Geli J, Nord B, Frisk T, Edström Elder E, EkströmTJ, et al. Deletions and altered expression of the RIZ1 tumour suppressorgene in 1p36 in pheochromocytomas and abdominal paragangliomas. Int JOncol. 2005; 26: 1385-1391. Ref: https://goo.gl/GCX4Yd
- Piao Z, Fang W, Malkhosyan S, Kim H, Horii A, etal. Frequent frameshift mutations of RIZ in sporadic gastrointestinal andendometrial carcinomas with microsatellite instability. Cancer Res. 2000;60: 4701-4704. Ref: https://goo.gl/W7x2i6
- Poetsch M, Dittberner T, Woenckhaus C. Frameshiftmutations of RIZ, but no point mutations in RIZ1 exons in malignantmelanomas with deletions in 1p36. Oncogene. 2002; 21: 3038-3042. Ref: https://goo.gl/aeV6mN
- Sakurada K, Furukawa T, Kato Y, Kayama, T, Huang,S. et al. RIZ, the retinoblastoma protein interacting zinc finger gene, ismutated in genetically unstable cancers of the pancreas, stomach, andcolorectum. Genes Chromosomes Cancer. 2001; 30: 207-211. Ref: https://goo.gl/RPyP9V
- Sasaki O, Meguro K, Tohmiya Y, Funato T,Shibahara, S, et al. Altered expression of retinoblastoma protein-interactingzinc finger gene, RIZ, in human leukaemia. Br J Haematol. 2002; 119: 940-948.Ref: https://goo.gl/7BPA7K
- Steele-Perkins G, Fang W, Yang XH, Van Gele M,Carling T, et al. Tumor formation and inactivation of RIZ1, an Rb-bindingmember of a nuclear protein-methyltransferase superfamily. Genes Dev. 2001;15: 2250-2262. Ref: https://goo.gl/LDUqkS
- Mzoughi S, Tan YX, Low D, Guccione E. The role ofPRDMs in cancer: one family, two sides. Curr Opin Genet Dev. 2016; 36: 83-91.Ref: https://goo.gl/aLyPC2
- Marmorstein R. Structure of SET domain proteins:a new twist on histone methylation.
- Trends Biochem Sci. 2003; 28: 59-62. Ref: https://goo.gl/fC7vwY
- Kim KC, Geng L, Huang S. Inactivation of ahistone methyltransferase by mutations in human cancers. Cancer Res. 2003;63: 7619-7623. Ref: https://goo.gl/aTL3SA
- Matsuo H, Goyama S, Kamikubo Y, Adachi S. Thesubtype-specific features of EVI1 and PRDM16 in acute myeloidleukemia. Haematologica. 2015; 100: e116-e117. Ref: https://goo.gl/X2BvyD
- Seale P, Bjork B, Yang W, Kajimura S, Chin S, etal. PRDM16 controls a brown fat/skeletal muscle switch.Nature. 2008; 454: 961-967. Ref: https://goo.gl/emNeao
- Yin H, Pasut A, Soleimani VD, Bentzinger CF,Antoun G, et al. MicroRNA-133 Controls Brown Adipose Determination inSkeletal Muscle Satellite Cells by Targeting Prdm16. Cell Metab. 2013; 17: 210-224. Ref: https://goo.gl/xU9ohE
- Deng Q, Huang S. PRDM5 is silenced in humancancers and has growth suppressive activities. Oncogene. 2004; 23: 4903-4910.Ref: https://goo.gl/fxFuV8
- Watanabe Y, Toyota M, Kondo Y, Suzuki H, Imai T,et al. PRDM5 identified as a target of epigenetic silencing in colorectaland gastric cancer. Clin Cancer Res. 2007; 13: 4786-4794. Ref: https://goo.gl/heTiF3
- Cheng HY, Chen XW, Cheng L, Liu YD, Lou G. DNAmethylation and carcinogenesis of PRDM5 in cervical cancer. J Cancer ResClin Oncol. 2010; 136: 1821-1825. Ref: https://goo.gl/uJwpLx
- Huang S. The retinoblastoma protein-interactingzinc finger gene RIZ in 1p36-linked cancers. Front Biosci. 1999; 4: D528-32.Ref: https://goo.gl/MEhmBJ
- Jiang Gl, Liu L, Buyse IM, Simon D, Huang S.Decreased RIZ1 expression but not RIZ2 in hepatoma and suppression ofhepatoma tumorigenicity by RIZ1. Int J cancer 1999; 83: 541-546. Ref: https://goo.gl/6tBdTJ
- Dong SW, Zhang YW, Chen Y, Wang S, Sun P, et al.PRDIBF1-RIZ domain of retinoblastoma protein-interacting zinc finger gene1 induces apoptosis and exerts anticancer activity in esophageal squamouscell carcinoma cells. Oncol Lett. 2015; 9: 341-346. Ref: https://goo.gl/Uv8Xc1
- Gazzerro P, Abbondanza C, Arcangelo A, Rossi M,Medici N, et al. Modulation of RIZ gene expression is associated toestradiol control of MCF-7 breast cancer cell proliferation. Exp Cell Res.2006; 312: 340-9. Ref: https://goo.gl/GzBzCz
- Rossi M, Abbondanza C, Arcangelo A, Gazzerro\ P, Medici N, Moncharmont B,
- Puca GA. The Zn-finger domain of RIZ protein promotes MCF-7 cell proliferation. Cancer
- Lett. 2004; 215: 229-37. Ref: https://goo.gl/Sj7TsU
- Medici N, Abbondanza C, Nigro V, Rossi V, PilusoG, et al. Identification of a DNA binding protein cooperating withestrogen receptor as RIZ (retinoblastoma interacting zinc finger protein).Biochem Biophys Res Commun. 1999; 264: 983-989. Ref: https://goo.gl/uN4qs9
- Abbondanza C, Medici N, Nigro V, Rossi V, GalloL, et al. The retinoblastoma-interacting zinc-finger protein RIZ is adownstream effector of estrogen action. Proc Natl Acad Sci. USA. 2000; 97:3130-3135. Ref: https://goo.gl/XoLXWf
- He L, Yu JX, Liu L, Buyse IM, Wang MS, et al.RIZ1, but not the alternative RIZ2 product of the same gene, isunderexpressed in breast cancer, and forced RIZ1 expression causes G2-Mcell cycle arrest and/or apoptosis. CancerRes. 1998; 58: 4238-4244.Ref: https://goo.gl/6xk6VN
- Koh-Stenta X, Joy J, Poulsen A, Li R, Tan Y, etal. Characterization of the histone methyltransferase PRDM9 usingbiochemical, biophysical and chemical biology techniques. Biochem J 2014; 461:323-334. Ref: https://goo.gl/M3WQge
- Li X, Wang J, Jiang Z, Guo F, Soloway PD, Zhao R.Role of PRDM16 and its PR domain in the epigenetic regulationof myogenic and adipogenic genes during transdifferentiation of C2C12cells. Gene 2015; 570: 191-198. Ref: https://goo.gl/ixp4Kr
- Harms MJ, Ishibashi J, Wang W, Lim HW, Goyama S, etal. Prdm16 is Required for the Maintenance of BrownAdipocyte Identity and Function in Adult Mice. Cell Metab. 2014; 19: 593-604. Ref: https://goo.gl/yTqTsM
- Burattini S, Ferri P, Battistelli M, Curci R,Luchetti F, et al. C2C12 murine myoblasts as a model of skeletal muscledevelopment: morpho-functional characterization. Eur J Histochem. 2004; 48:223-233. Ref: https://goo.gl/MwDgzE
- Cheedipudi S, Puri D, Saleh A, Gala HP, Rumman M,et al. A fine balance: epigenetic control of cellular quiescence by thetumor suppressor PRDM2/RIZ at a bivalent domain in the cyclin a gene.Nucleic Acids Res. 2015; 43:6236-56. Ref: https://goo.gl/B3UYF7
- Sabourin LA, Rudnicki M.A. The molecularregulation of myogenesis. Clin Genet. 2000; 57: 16-25. Ref: https://goo.gl/yHVw8p
- Morano A, Angrisano T, Russo G, Landi R, PezoneA, et al. Targeted DNA methylation by homology-directed repair inmammalian cells. Transcription reshapes methylation on the repaired gene.Nucleic Acids Res. 2014; 42: 804-821. Ref: https://goo.gl/NUDMA3
- Pfaffl MW. A new mathematical model for relativequantification in real-time RT-PCR. Nucleic Acids Res. 2001; 29: 45. Ref: https://goo.gl/ABUUGz
- Laemmli UK. Cleavage of structural proteinsduring the assembly of the head of bacteriophage T4. Nature. 1970; 227:680-685. Ref: https://goo.gl/jS8Gfa
- Crescenzi M, Crouch DH, Tatò F. Transformation bymyc prevents fusion but not biochemical differentiation of C2C12myoblasts: mechanisms of phenotypic correction in mixed culture withnormal cells. J Cell Biol. 1994;125: 1137-1145. Ref: https://goo.gl/bNkD5j
- Mal A, Harter ML. MyoD is functionally linked tothe silencing of a muscle-specific regulatory gene prior to skeletalmyogenesis. Proc Natl. Acad Sci. 2003; 100: 1735-1739. Ref: https://goo.gl/cdc72F
- Gazzerro P, Bontempo P, Schiavone EM, AbbondanzaC, Moncharmont B, et al. Differentiation of myeloid cell lines correlateswith a selective expression of RIZ protein. Mol Med. 2001; 7: 552-560. Ref: https://goo.gl/PJDGtc
- Abbondanza C, Nigris F, Rosa C, Rossiello R, PucaGA, et al. Silencing of YY1 downregulates RIZ1 promoter in humanosteosarcoma. Oncol Res. 2008;17: 33-41. Ref: https://goo.gl/Cuwz43
- Mal AK. Histone methyltransferase Suv39h1 represses MyoD-stimulated myogenic differentiation. EMBO J. 2006; 25: 3323-3334. Ref: https://goo.gl/jr1Sse